cTWAS_analysis_for_RA
Jing Gu
2023-08-16
Last updated: 2023-08-16
Checks: 6 1
Knit directory: m6A_in_disease_genetics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230331) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/projects/m6A_in_disease_genetics/code/ctwas/ctwas_config_b37.R | code/ctwas/ctwas_config_b37.R |
| ~/projects/m6A_in_disease_genetics/code/ctwas/qiansheng/locus_plot.R | code/ctwas/qiansheng/locus_plot.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 08eaf44. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/m6A_switch_to_disease_h2g.nb.html
Ignored: data/plots/
Untracked files:
Untracked: HMGCR_locus_gene_tracks.pdf
Untracked: Rplots.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/IBD_E_S_m6A.Rmd
Untracked: analysis/IBD_E_S_m6A_output.Rmd
Untracked: analysis/LDL_E_S_m6A.Rmd
Untracked: analysis/LDL_m6A_output.Rmd
Untracked: analysis/RA_m6A_output.Rmd
Untracked: analysis/WhiteBlood_WholeBlood_E_M.Rmd
Untracked: analysis/identify_m6A_mechanisms_with_finemapping.Rmd
Untracked: analysis/lymph_m6A_output.Rmd
Untracked: analysis/pre_weights_m6AQTL.txt
Untracked: analysis/rbc_E_S_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output_hg19.Rmd
Untracked: analysis/wbc_E_S_m6A_output.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: code/check_double_strand.ipynb
Untracked: code/check_double_strand_v2.ipynb
Untracked: code/ctwas/
Untracked: code/figure/
Untracked: code/learn_gviz.Rmd
Untracked: code/learn_gviz.html
Untracked: code/learn_gviz.nb.html
Untracked: code/m6AQTL_finemapping.Rmd
Untracked: code/summary_TWAS_coloc_m6A_2023.Rmd
Untracked: code/test_gviz.ipynb
Untracked: code/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: data/.ipynb_checkpoints/
Untracked: data/ADCY7_gwas_input.tsv
Untracked: data/ADCY7_qtl_input.tsv
Untracked: data/Allergy_full_coloc.txt
Untracked: data/Asthma_full_coloc.txt
Untracked: data/CAD_full_coloc.txt
Untracked: data/Eosinophil_count_full_coloc.txt
Untracked: data/GSE125377_jointPeakReadCount.txt
Untracked: data/HMGCR_ctwas_dat.Rd
Untracked: data/IBD_full_coloc.txt
Untracked: data/JointPeaks.bed
Untracked: data/Li2022_dsRNAs.xlsx
Untracked: data/Lupus_full_coloc.txt
Untracked: data/RA_full_coloc.txt
Untracked: data/TABLE1_hg19.txt
Untracked: data/TABLE1_hg19.txt.zip
Untracked: data/__MACOSX/
Untracked: data/coloc_blood_traits.csv
Untracked: data/crohns_disease_full_coloc.txt
Untracked: data/edit_sites_and_GE_neg_correlated.txt
Untracked: data/edit_sites_and_GE_pos_correlated.txt
Untracked: data/features
Untracked: data/human_EERs.csv
Untracked: data/human_EERs.txt
Untracked: data/lymph_full_coloc.txt
Untracked: data/m6A_TWAS_results.csv
Untracked: data/m6a_TWAS_genes.txt
Untracked: data/m6a_joint_calling_peaks.csv
Untracked: data/nasser_2021_ABC_IBD_genes.txt
Untracked: data/nat_sense_pairs.csv
Untracked: data/plt_full_coloc.txt
Untracked: data/rbc_full_coloc.txt
Untracked: data/rdw_full_coloc.txt
Untracked: data/reported_AS_targets_S1.txt
Untracked: data/reported_AS_wanowska.txt
Untracked: data/sig_coloc_results/
Untracked: data/test_locuscomparer.pdf
Untracked: data/ulcerative_colitis_full_coloc.txt
Untracked: data/wbc_full_coloc.txt
Untracked: data/zhao_silver_genes.csv
Untracked: output/.ipynb_checkpoints/
Untracked: output/HMGCR_gene_track_plot.pdf
Untracked: output/HMGCR_locus_plot.pdf
Untracked: output/all_m6a_sites_with_cisNATs.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.3.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.5.csv
Untracked: output/all_m6a_sites_with_paired_cis_NATs.csv
Untracked: output/fine_mapped_m6AQTLs_TWAS_genes_highPP4.rds
Untracked: output/gene_summary.csv
Untracked: output/immune_related_m6A_targets.csv
Untracked: output/m6aQTL_dsRNAs_PPP2R3C_PRORP.pdf
Untracked: output/m6a_peaks_nearby_dsRNAs.csv
Untracked: output/m6a_sites_near_all_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_coloc.csv
Untracked: output/m6a_sites_near_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_twas_summary.csv
Untracked: output/m6a_sites_overlapping_NAT_twas.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_coloc.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_twas.csv
Untracked: output/m6a_sites_overlapping_dsRegions.csv
Untracked: output/m6a_sites_overlapping_dsRegions_coloc.csv
Untracked: output/negatively_correlated_genes.txt
Untracked: output/postively_correlated_genes.txt
Untracked: output/rs1806261_RABEP1-NUP88_focused_locusview.pdf
Untracked: output/rs1806261_RABEP1-NUP88_locusview.pdf
Untracked: output/rs3177647_MAPKAPK5-AS1-MAPKAPK5_locusview.pdf
Untracked: output/rs3204541_DDX55-EIF2B1_locusview.pdf
Untracked: output/rs7184802_ADCY7-BRD7_locusview.pdf
Untracked: output/rs7184802_ADCY7_locuscompare.pdf
Untracked: output/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: output/twas_genes_PP4_0.5_blood_traits_trackplots.pdf
Untracked: output/twas_m6a_sites_with_all_cisNATs.RDS
Untracked: output/twas_m6a_sites_with_cisNATs_range.RDS
Untracked: output/twas_m6a_sites_with_the_nearest_cisNAT.RDS
Untracked: twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Unstaged changes:
Modified: analysis/m6A_switch_to_disease_h2g.Rmd
Modified: analysis/wbc_m6A_output.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/RA_m6A_output_hg19.Rmd)
and HTML (docs/RA_m6A_output_hg19.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 08eaf44 | Jing Gu | 2023-08-16 | run ctwas for multiple traits |
Load ctwas results
# top 1 method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "recover",
recover_strand_ambig_wgt = T
# lasso/elastic-net method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "none",
recover_strand_ambig_wgt = FGWAS: UK Biobank GWAS summary statistics - European individuals
Weights: FUSION weights using top1, lasso, or elastic-net models were converted into PredictDB format and were not needed to do scaling when running ctwas.
Check convergence of parameters
cTWAS analysis on m6A alone
[1] "Check convergence for the top1 model:"
[1] "Table of group size:"
SNP gene
7546780 887
SNP gene
estimated_group_prior 0.0001245 5.404e-04
estimated_group_prior_var 2.6814184 3.340e-01
estimated_group_pve 0.0074711 4.749e-07
attributable_group_pve 0.9999364 6.357e-05$top1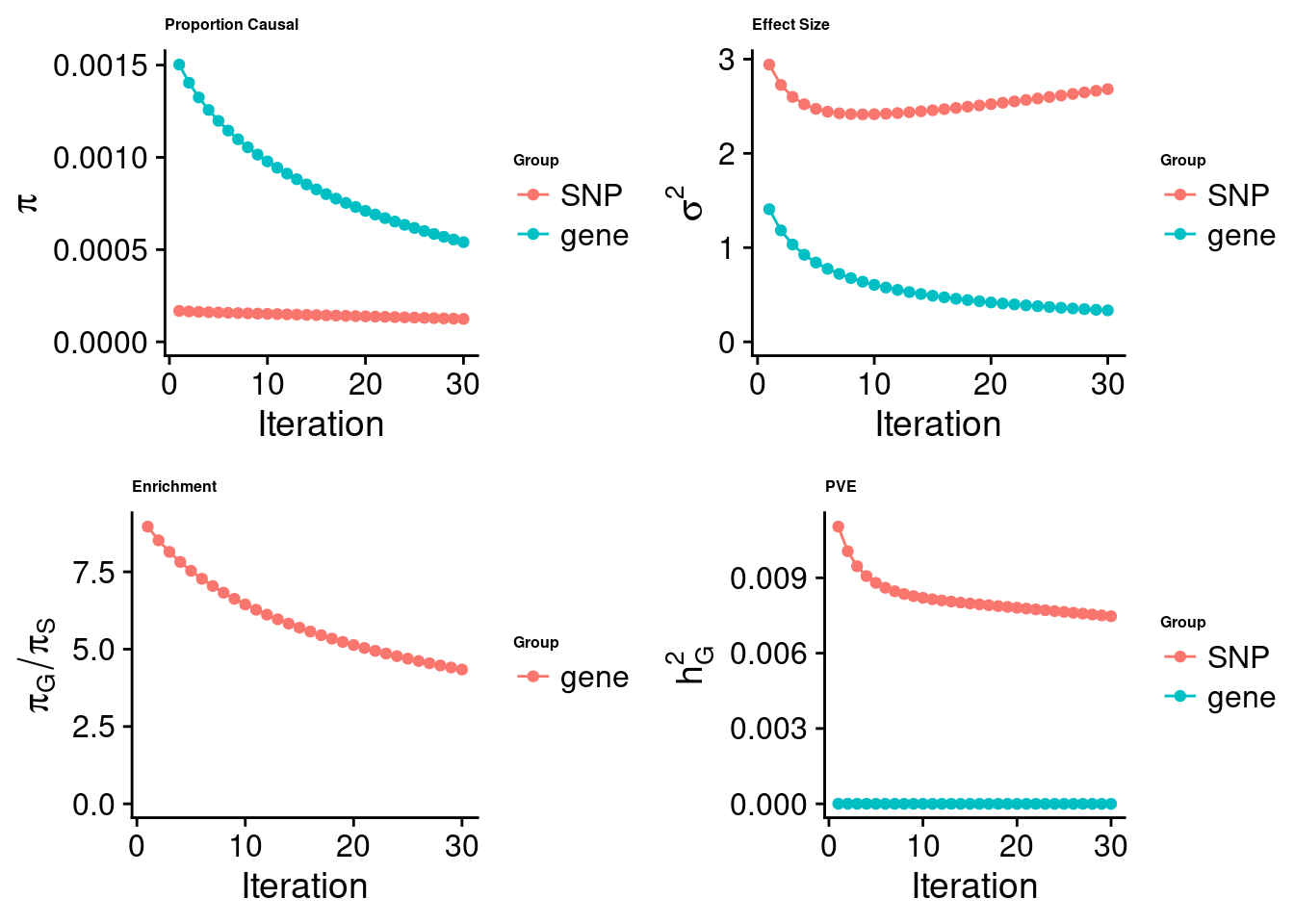
Joint analysis of expression, splicing and m6A
[1] "Check convergence for the top1 model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.0000 918.0000
group_size 7.547e+06 1928.0000 2122.0000 887.0000
percent_of_overlaps 8.094e-01 0.9616 0.9685 0.9662
SNP eQTL sQTL m6AQTL
estimated_group_prior 0.000122 7.439e-04 0.0028751 5.816e-04
estimated_group_prior_var 2.677861 3.618e-01 5.3000652 3.193e-01
estimated_group_pve 0.007312 1.539e-06 0.0000959 4.886e-07
attributable_group_pve 0.986784 2.077e-04 0.0129426 6.594e-05
[1] "Check convergence for the lasso model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 7.547e+06 1998.0000 2180.000 911.0000
percent_of_overlaps 8.094e-01 0.9965 0.995 0.9924
SNP eQTL sQTL m6AQTL
estimated_group_prior 0.0001062 3.235e-04 0.0072035 2.883e-04
estimated_group_prior_var 2.9452387 2.699e-01 3.4938813 4.698e-01
estimated_group_pve 0.0069980 5.175e-07 0.0001627 3.659e-07
attributable_group_pve 0.9771539 7.227e-05 0.0227228 5.110e-05$top1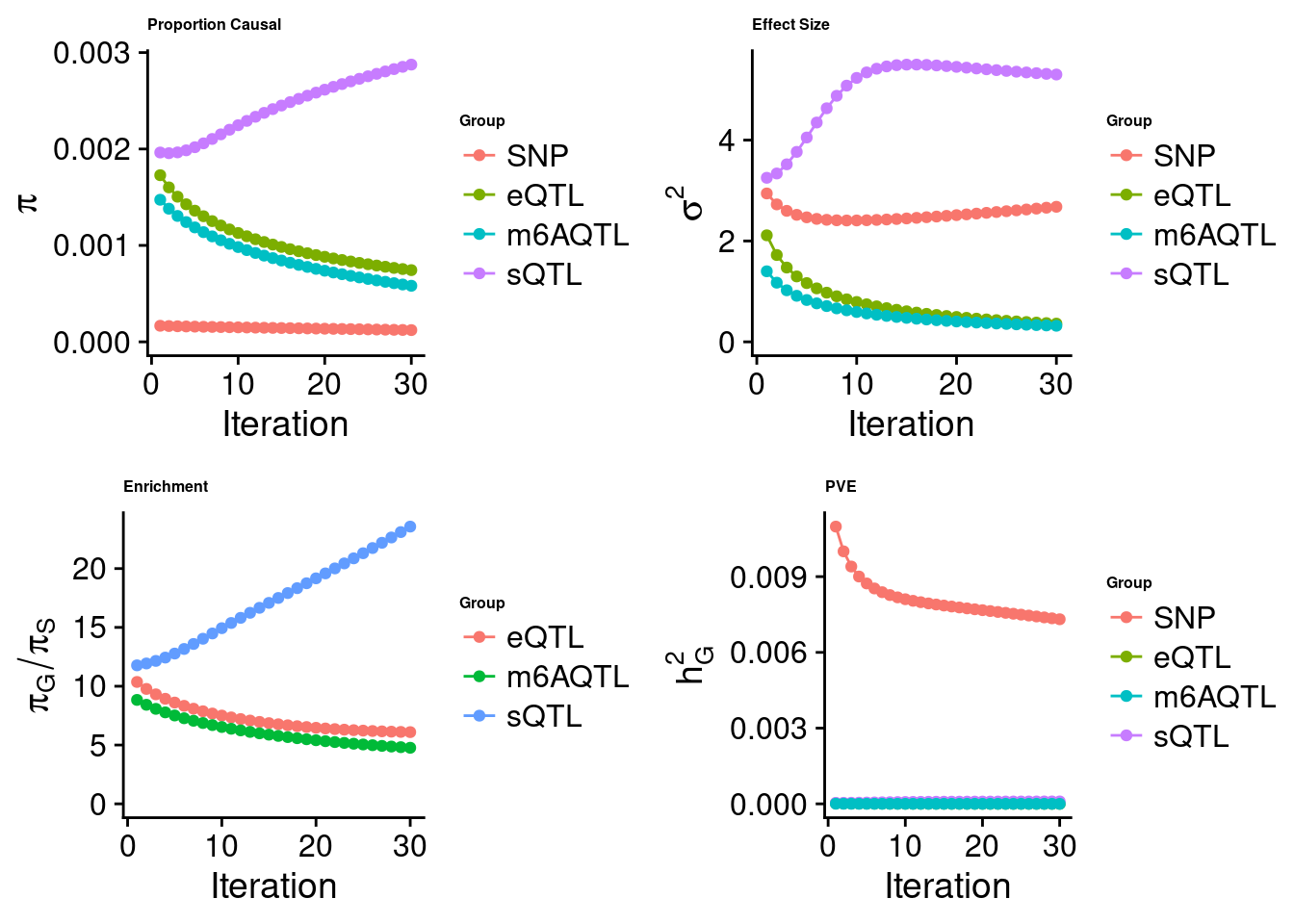
$lasso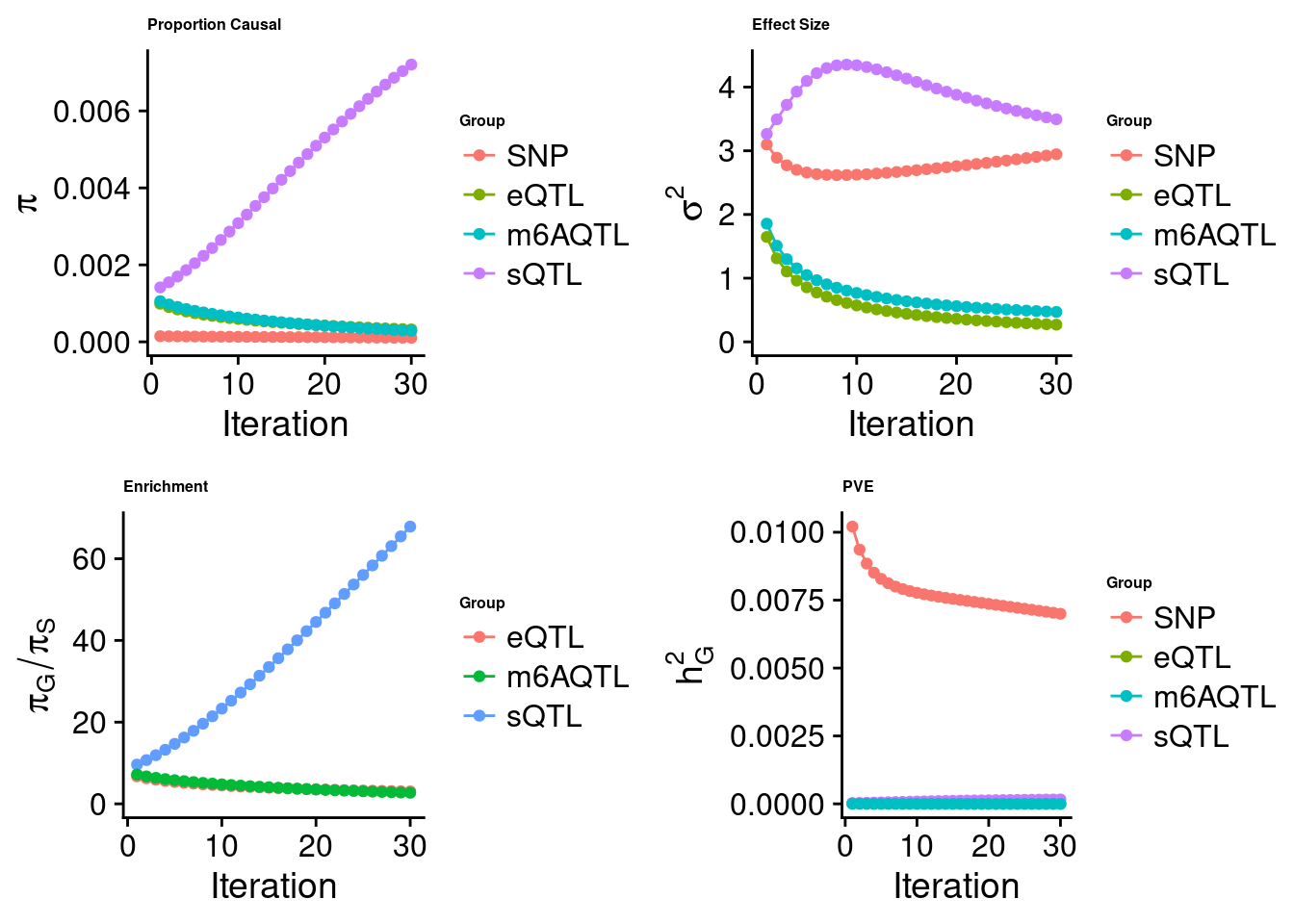
cTWAS results for individual analysis with m6A
top1 model
[1] genename region_tag susie_pip z
<0 rows> (or 0-length row.names)Summing up PIPs for m6A peaks located in the same gene
Top m6A PIPs by genes
# A tibble: 0 × 2
# ℹ 2 variables: genename <chr>, total_susie_pip <dbl>cTWAS results for joint analysis using a lasso model
Top m6A modification pip
Top expression/splicing/m6A units
For m6A or splicing QTLs, they are assigned to the nearest genes (m6A needs to be confirmed with Kevin).
Top SNPs or genes with PIP > 0.6
$eQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)
$m6AQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)
$sQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)Top m6A modification pip
ZKSCAN5: RNA Polymerase II Cis-Regulatory Region Sequence-Specific DNA Binding (GO:0000978). THEMIS2 is involved in the biological process T Cell Receptor Signaling Pathway (GO:0050852). BANF: DNA binding factor|Regulation Of Innate Immune Response (GO:0045088). TRIT1 has the molecular function of Catalytic Activity, Acting On A tRNA (GO:0140101). TRIT1 is involved in the biological process RNA Modification (GO:0009451). S1PR2 is involved in the biological process Regulation Of Cell Population Proliferation (GO:0042127). WAC has the molecular function of RNA Polymerase II Complex Binding (GO:0000993). CD320 is involved in the biological process Regulation Of B Cell Proliferation (GO:0030888).
genename region_tag susie_pip z
1 ZNF282 7_92 0.005477 -3.516
2 DAPP1 4_68 0.004313 -3.427
3 THEMIS2 1_19 0.003782 -2.742
4 REXO4 9_70 0.003372 -2.869
5 REXO4 9_70 0.003312 2.843
6 TNIP2 4_4 0.003308 -2.502
7 SURF4 9_70 0.003269 -2.817
8 SLC25A33 1_7 0.003150 -2.408
9 TCTN3 10_61 0.003065 -2.801
10 C12orf45 12_63 0.002905 2.494Summing up PIPs for m6A peaks located in the same gene
Top 10 m6A PIPs by genes
# A tibble: 818 × 2
genename total_susie_pip
<chr> <dbl>
1 PCNT 0.00989
2 PARP14 0.00711
3 REXO4 0.00668
4 CENPF 0.00573
5 ZNF282 0.00548
6 NSUN4 0.00527
7 MTERF4 0.00514
8 ICOSLG 0.00511
9 SUGP2 0.00510
10 AHSA2 0.00482
# ℹ 808 more rowsTop splicing PIPs
Some loci contain variants in the same credible set but having opposite z scores. For instance, the predicted splicing levels of two introns of CNN2 based on the same variant (position=1038445) have opposite associations with traits. Is this variant more likely to affect traits by altering the splicing levels of both transcripts, rather than one of them since they have equal PIP?
peak_id genename pos region_tag susie_pip z
1 chr7:75625917-75633076 STYXL1 75588366 7_48 0.2123 3.632
2 chr14:78154160-78161081 ALKBH1 78088985 14_36 0.1926 -3.128
3 chr19:38872868-38873893 PSMD8 38773966 19_27 0.1871 -2.865
4 chr20:57248767-57266780 NPEPL1 57165617 20_34 0.1817 2.817
5 chr11:71721900-71723447 NUMA1 71626089 11_40 0.1762 4.593
6 chr19:13886427-13888866 C19orf53 13938903 19_11 0.1532 -2.777
7 chr2:10583439-10583616 ODC1 10580967 2_7 0.1493 2.642
8 chr15:74711293-74717716 SEMA7A 74630623 15_35 0.1446 2.880
9 chr5:96075822-96076970 CAST 96057891 5_57 0.1444 3.226
10 chr5:96076487-96076970 CAST 96071780 5_57 0.1444 -3.225Summing up PIPs for spliced introns located in the same gene
Top 10 splicing PIPs by genes
# A tibble: 10 × 2
genename total_susie_pip
<chr> <dbl>
1 STYXL1 0.549
2 CAST 0.471
3 ATP5PO 0.402
4 NUMA1 0.385
5 TRAF1 0.347
6 WARS1 0.346
7 CCT7 0.324
8 CD46 0.321
9 NDUFB1 0.289
10 OAS1 0.285Top genes by combined PIP
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
2871 STYXL1 0.551 0.001569 0.5492 0.000000 7_48
414 CAST 0.471 0.000000 0.4710 0.000000 5_57
252 ATP5PO 0.404 0.002052 0.4016 0.000000 21_15
2092 NUMA1 0.385 0.000000 0.3852 0.000000 11_40
3240 WARS1 0.348 0.001807 0.3460 0.000000 14_52
3072 TRAF1 0.347 0.000000 0.3471 0.000000 9_63
454 CCT7 0.324 0.000000 0.3237 0.000000 2_48
471 CD46 0.321 0.000000 0.3213 0.000000 1_107
2006 NDUFB1 0.289 0.000000 0.2892 0.000000 14_47
2107 OAS1 0.286 0.000000 0.2846 0.001857 12_68
1701 LITAF 0.255 0.000000 0.2546 0.000000 16_12
1784 MCM3 0.255 0.000000 0.2546 0.000000 6_39
528 CFLAR 0.250 0.000000 0.2478 0.002544 2_119
1304 FANCL 0.235 0.000000 0.2348 0.000000 2_39
1572 IMMP1L 0.233 0.000000 0.2329 0.000000 11_21
2366 PSMD8 0.223 0.000000 0.2232 0.000000 19_27
1552 IFI44L 0.220 0.000000 0.2188 0.001382 1_48
2503 RMDN1 0.213 0.000000 0.2126 0.000000 8_62
138 ALKBH1 0.210 0.000000 0.2103 0.000000 14_36
1790 MCOLN2 0.210 0.001845 0.2082 0.000000 1_52
1957 MYO1G 0.208 0.000000 0.2079 0.000000 7_33
1608 ITPA 0.201 0.001845 0.1995 0.000000 20_3
346 C11orf24 0.199 0.000000 0.1988 0.000000 11_38
349 C12orf73 0.199 0.000000 0.1994 0.000000 12_62
2788 SNX2 0.192 0.002529 0.1893 0.000000 5_74
2700 SLC1A3 0.191 0.000000 0.1909 0.000000 5_25
1680 LBP 0.190 0.000000 0.1896 0.000000 20_23
469 CD40 0.188 0.000000 0.1878 0.000000 20_28
3172 UBL7 0.188 0.000000 0.1883 0.000000 15_35
359 C19orf53 0.187 0.000000 0.1856 0.001281 19_11
2071 NSUN4 0.186 0.001888 0.1786 0.005265 1_29
572 COA1 0.182 0.000000 0.1818 0.000000 7_33
2058 NPEPL1 0.182 0.000000 0.1817 0.000000 20_34
2800 SP140 0.177 0.000000 0.1771 0.000000 2_135
2607 RWDD3 0.176 0.001675 0.1728 0.001396 1_58
2729 SLC3A2 0.175 0.000000 0.1752 0.000000 11_35
2312 PPIL3 0.172 0.001672 0.1687 0.001147 2_119
2246 PLEKHB2 0.169 0.000000 0.1692 0.000000 2_78
1968 NADSYN1 0.167 0.001343 0.1661 0.000000 11_40
3013 TMEM230 0.167 0.000000 0.1672 0.000000 20_4Loading required package: gridWarning: replacing previous import 'utils::download.file' by
'restfulr::download.file' when loading 'rtracklayer'Locus plots for specific examples
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
1509 HMGCR 0.002 0 0 0.001878 5_44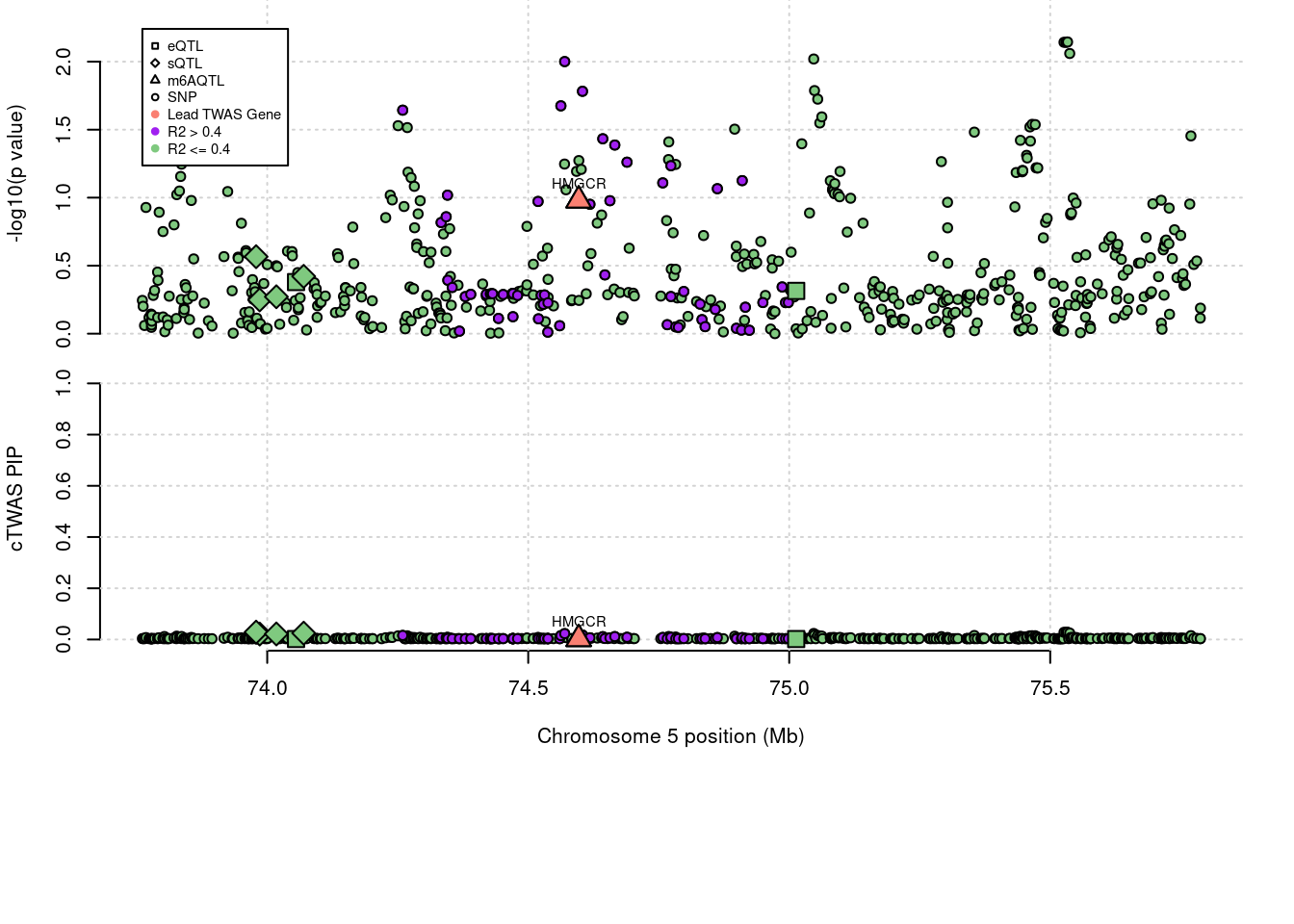
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.52.0 Gviz_1.40.1 cowplot_1.1.1
[4] ggplot2_3.4.3 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[7] IRanges_2.30.1 S4Vectors_0.34.0 BiocGenerics_0.42.0
[10] ctwas_0.1.38 dplyr_1.1.2 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 deldir_1.0-6
[3] rjson_0.2.21 rprojroot_2.0.3
[5] biovizBase_1.44.0 htmlTable_2.4.0
[7] XVector_0.36.0 base64enc_0.1-3
[9] fs_1.6.3 dichromat_2.0-0.1
[11] rstudioapi_0.15.0 farver_2.1.1
[13] bit64_4.0.5 AnnotationDbi_1.58.0
[15] fansi_1.0.4 xml2_1.3.3
[17] codetools_0.2-18 logging_0.10-108
[19] cachem_1.0.8 knitr_1.39
[21] Formula_1.2-4 jsonlite_1.8.7
[23] Rsamtools_2.12.0 cluster_2.1.3
[25] dbplyr_2.3.3 png_0.1-7
[27] compiler_4.2.0 httr_1.4.6
[29] backports_1.4.1 lazyeval_0.2.2
[31] Matrix_1.6-1 fastmap_1.1.1
[33] cli_3.6.1 later_1.3.0
[35] htmltools_0.5.2 prettyunits_1.1.1
[37] tools_4.2.0 gtable_0.3.3
[39] glue_1.6.2 GenomeInfoDbData_1.2.8
[41] rappdirs_0.3.3 Rcpp_1.0.11
[43] Biobase_2.56.0 jquerylib_0.1.4
[45] vctrs_0.6.3 Biostrings_2.64.0
[47] rtracklayer_1.56.0 iterators_1.0.14
[49] xfun_0.30 stringr_1.5.0
[51] ps_1.7.0 lifecycle_1.0.3
[53] ensembldb_2.20.2 restfulr_0.0.14
[55] XML_3.99-0.14 getPass_0.2-2
[57] zlibbioc_1.42.0 scales_1.2.1
[59] BSgenome_1.64.0 VariantAnnotation_1.42.1
[61] ProtGenerics_1.28.0 hms_1.1.3
[63] promises_1.2.0.1 MatrixGenerics_1.8.0
[65] parallel_4.2.0 SummarizedExperiment_1.26.1
[67] AnnotationFilter_1.20.0 RColorBrewer_1.1-3
[69] yaml_2.3.5 curl_5.0.2
[71] memoise_2.0.1 gridExtra_2.3
[73] sass_0.4.1 rpart_4.1.16
[75] latticeExtra_0.6-30 stringi_1.7.12
[77] RSQLite_2.3.1 highr_0.9
[79] BiocIO_1.6.0 foreach_1.5.2
[81] checkmate_2.1.0 GenomicFeatures_1.48.4
[83] filelock_1.0.2 BiocParallel_1.30.3
[85] rlang_1.1.1 pkgconfig_2.0.3
[87] matrixStats_0.62.0 bitops_1.0-7
[89] evaluate_0.15 lattice_0.20-45
[91] htmlwidgets_1.5.4 GenomicAlignments_1.32.0
[93] labeling_0.4.2 bit_4.0.5
[95] processx_3.8.0 tidyselect_1.2.0
[97] magrittr_2.0.3 R6_2.5.1
[99] generics_0.1.3 Hmisc_5.1-0
[101] DelayedArray_0.22.0 DBI_1.1.3
[103] pgenlibr_0.3.6 pillar_1.9.0
[105] whisker_0.4 foreign_0.8-82
[107] withr_2.5.0 KEGGREST_1.36.2
[109] RCurl_1.98-1.7 nnet_7.3-17
[111] tibble_3.2.1 crayon_1.5.2
[113] interp_1.1-4 utf8_1.2.3
[115] BiocFileCache_2.4.0 rmarkdown_2.14
[117] jpeg_0.1-10 progress_1.2.2
[119] data.table_1.14.8 blob_1.2.4
[121] callr_3.7.3 git2r_0.30.1
[123] digest_0.6.33 httpuv_1.6.5
[125] munsell_0.5.0 bslib_0.3.1