cTWAS_analysis_for_red_blood_counts
Jing Gu
2023-08-16
Last updated: 2023-08-16
Checks: 6 1
Knit directory: m6A_in_disease_genetics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230331) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/projects/m6A_in_disease_genetics/code/ctwas/ctwas_config_b37.R | code/ctwas/ctwas_config_b37.R |
| ~/projects/m6A_in_disease_genetics/code/ctwas/qiansheng/locus_plot.R | code/ctwas/qiansheng/locus_plot.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version a1e2443. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/m6A_switch_to_disease_h2g.nb.html
Ignored: data/plots/
Untracked files:
Untracked: HMGCR_locus_gene_tracks.pdf
Untracked: Rplots.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/IBD_E_S_m6A.Rmd
Untracked: analysis/IBD_E_S_m6A_output.Rmd
Untracked: analysis/LDL_E_S_m6A.Rmd
Untracked: analysis/LDL_m6A_output.Rmd
Untracked: analysis/RA_m6A_output.Rmd
Untracked: analysis/WhiteBlood_WholeBlood_E_M.Rmd
Untracked: analysis/identify_m6A_mechanisms_with_finemapping.Rmd
Untracked: analysis/lymph_m6A_output.Rmd
Untracked: analysis/pre_weights_m6AQTL.txt
Untracked: analysis/rbc_E_S_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output.Rmd
Untracked: analysis/wbc_E_S_m6A_output.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: code/annotating_fine-mapped_m6A_QTLs.Rmd
Untracked: code/check_double_strand.ipynb
Untracked: code/check_double_strand_v2.ipynb
Untracked: code/ctwas/
Untracked: code/figure/
Untracked: code/learn_gviz.Rmd
Untracked: code/learn_gviz.html
Untracked: code/learn_gviz.nb.html
Untracked: code/m6AQTL_finemapping.Rmd
Untracked: code/summary_TWAS_coloc_m6A_2023.Rmd
Untracked: code/test_gviz.ipynb
Untracked: code/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: data/.ipynb_checkpoints/
Untracked: data/ADCY7_gwas_input.tsv
Untracked: data/ADCY7_qtl_input.tsv
Untracked: data/Allergy_full_coloc.txt
Untracked: data/Asthma_full_coloc.txt
Untracked: data/CAD_full_coloc.txt
Untracked: data/Eosinophil_count_full_coloc.txt
Untracked: data/GSE125377_jointPeakReadCount.txt
Untracked: data/HMGCR_ctwas_dat.Rd
Untracked: data/IBD_full_coloc.txt
Untracked: data/JointPeaks.bed
Untracked: data/Li2022_dsRNAs.xlsx
Untracked: data/Lupus_full_coloc.txt
Untracked: data/RA_full_coloc.txt
Untracked: data/TABLE1_hg19.txt
Untracked: data/TABLE1_hg19.txt.zip
Untracked: data/__MACOSX/
Untracked: data/coloc_blood_traits.csv
Untracked: data/crohns_disease_full_coloc.txt
Untracked: data/edit_sites_and_GE_neg_correlated.txt
Untracked: data/edit_sites_and_GE_pos_correlated.txt
Untracked: data/features
Untracked: data/human_EERs.csv
Untracked: data/human_EERs.txt
Untracked: data/lymph_full_coloc.txt
Untracked: data/m6A_TWAS_results.csv
Untracked: data/m6a_TWAS_genes.txt
Untracked: data/m6a_joint_calling_peaks.csv
Untracked: data/nasser_2021_ABC_IBD_genes.txt
Untracked: data/nat_sense_pairs.csv
Untracked: data/plt_full_coloc.txt
Untracked: data/rbc_full_coloc.txt
Untracked: data/rdw_full_coloc.txt
Untracked: data/reported_AS_targets_S1.txt
Untracked: data/reported_AS_wanowska.txt
Untracked: data/sig_coloc_results/
Untracked: data/test_locuscomparer.pdf
Untracked: data/ulcerative_colitis_full_coloc.txt
Untracked: data/wbc_full_coloc.txt
Untracked: data/zhao_silver_genes.csv
Untracked: output/.ipynb_checkpoints/
Untracked: output/HMGCR_gene_track_plot.pdf
Untracked: output/HMGCR_locus_plot.pdf
Untracked: output/all_m6a_sites_with_cisNATs.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.3.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.5.csv
Untracked: output/all_m6a_sites_with_paired_cis_NATs.csv
Untracked: output/fine_mapped_m6AQTLs_TWAS_genes_highPP4.rds
Untracked: output/gene_summary.csv
Untracked: output/immune_related_m6A_targets.csv
Untracked: output/m6aQTL_dsRNAs_PPP2R3C_PRORP.pdf
Untracked: output/m6a_peaks_nearby_dsRNAs.csv
Untracked: output/m6a_sites_near_all_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_coloc.csv
Untracked: output/m6a_sites_near_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_twas_summary.csv
Untracked: output/m6a_sites_overlapping_NAT_twas.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_coloc.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_twas.csv
Untracked: output/m6a_sites_overlapping_dsRegions.csv
Untracked: output/m6a_sites_overlapping_dsRegions_coloc.csv
Untracked: output/negatively_correlated_genes.txt
Untracked: output/postively_correlated_genes.txt
Untracked: output/rs1806261_RABEP1-NUP88_focused_locusview.pdf
Untracked: output/rs1806261_RABEP1-NUP88_locusview.pdf
Untracked: output/rs3177647_MAPKAPK5-AS1-MAPKAPK5_locusview.pdf
Untracked: output/rs3204541_DDX55-EIF2B1_locusview.pdf
Untracked: output/rs7184802_ADCY7-BRD7_locusview.pdf
Untracked: output/rs7184802_ADCY7_locuscompare.pdf
Untracked: output/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: output/twas_genes_PP4_0.5_blood_traits_trackplots.pdf
Untracked: output/twas_m6a_sites_with_all_cisNATs.RDS
Untracked: output/twas_m6a_sites_with_cisNATs_range.RDS
Untracked: output/twas_m6a_sites_with_the_nearest_cisNAT.RDS
Untracked: twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Unstaged changes:
Modified: analysis/m6A_switch_to_disease_h2g.Rmd
Modified: analysis/wbc_m6A_output.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/rbc_m6A_output_hg19.Rmd)
and HTML (docs/rbc_m6A_output_hg19.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | a1e2443 | Jing Gu | 2023-08-16 | added rbc |
Load ctwas results
# top 1 method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "recover",
recover_strand_ambig_wgt = T
# lasso/elastic-net method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "none",
recover_strand_ambig_wgt = FGWAS: UK Biobank GWAS summary statistics - European individuals
Weights: FUSION weights using top1, lasso, or elastic-net models were converted into PredictDB format and were not needed to do scaling when running ctwas.
Check convergence of parameters
cTWAS analysis on m6A alone
[1] "Check convergence for the top1 model:"
[1] "Table of group size:"
SNP gene
8713250 888
SNP gene
estimated_group_prior 2.452e-04 2.534e-08
estimated_group_prior_var 2.551e+01 1.357e+01
estimated_group_pve 1.555e-01 8.711e-10
attributable_group_pve 1.000e+00 5.601e-09
[1] "Check convergence for the lasso model:"
[1] "Table of group size:"
SNP gene
8713250 912
SNP gene
estimated_group_prior 2.389e-04 1.809e-06
estimated_group_prior_var 2.384e+01 2.900e+01
estimated_group_pve 1.416e-01 1.365e-07
attributable_group_pve 1.000e+00 9.643e-07$top1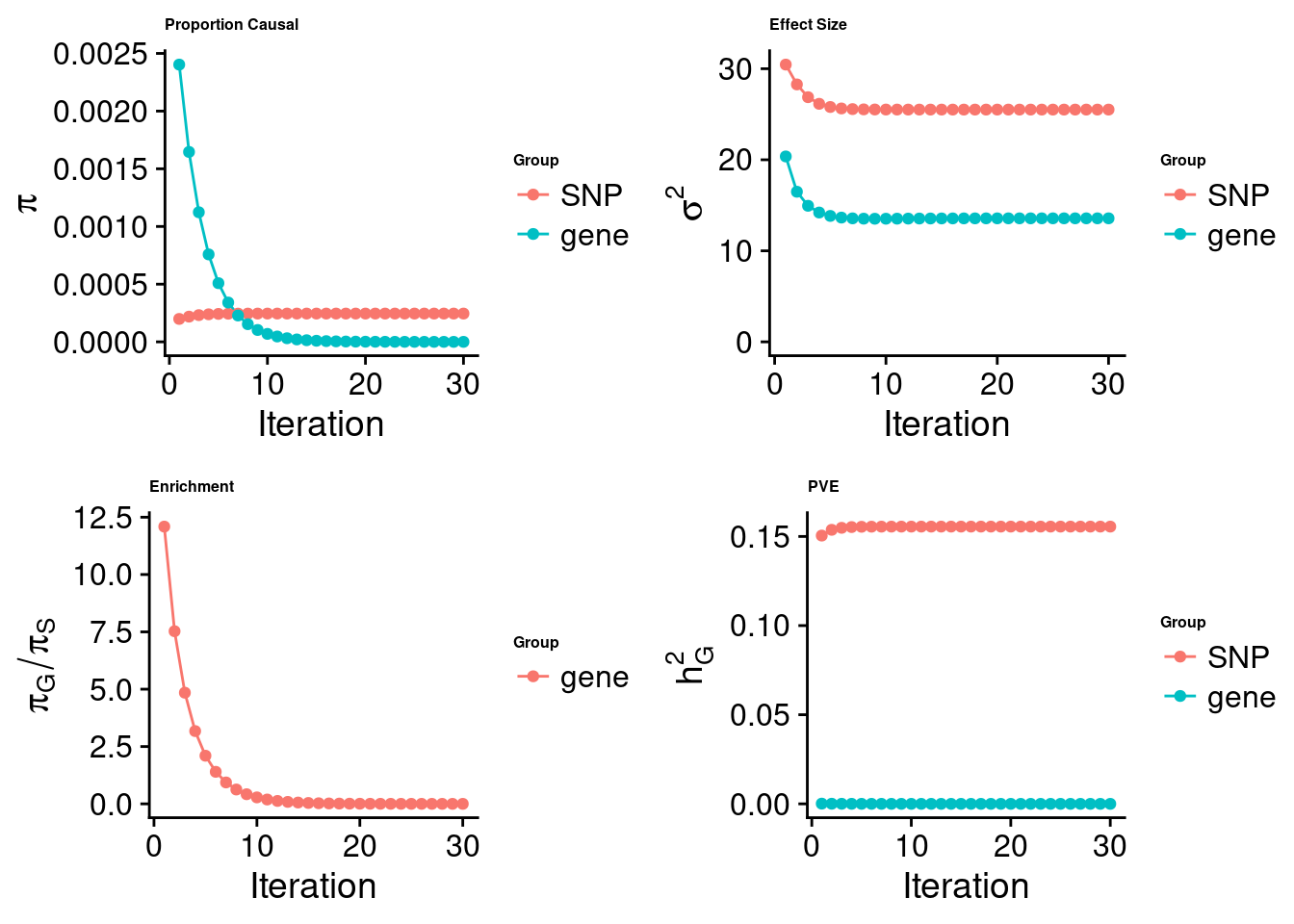
$lasso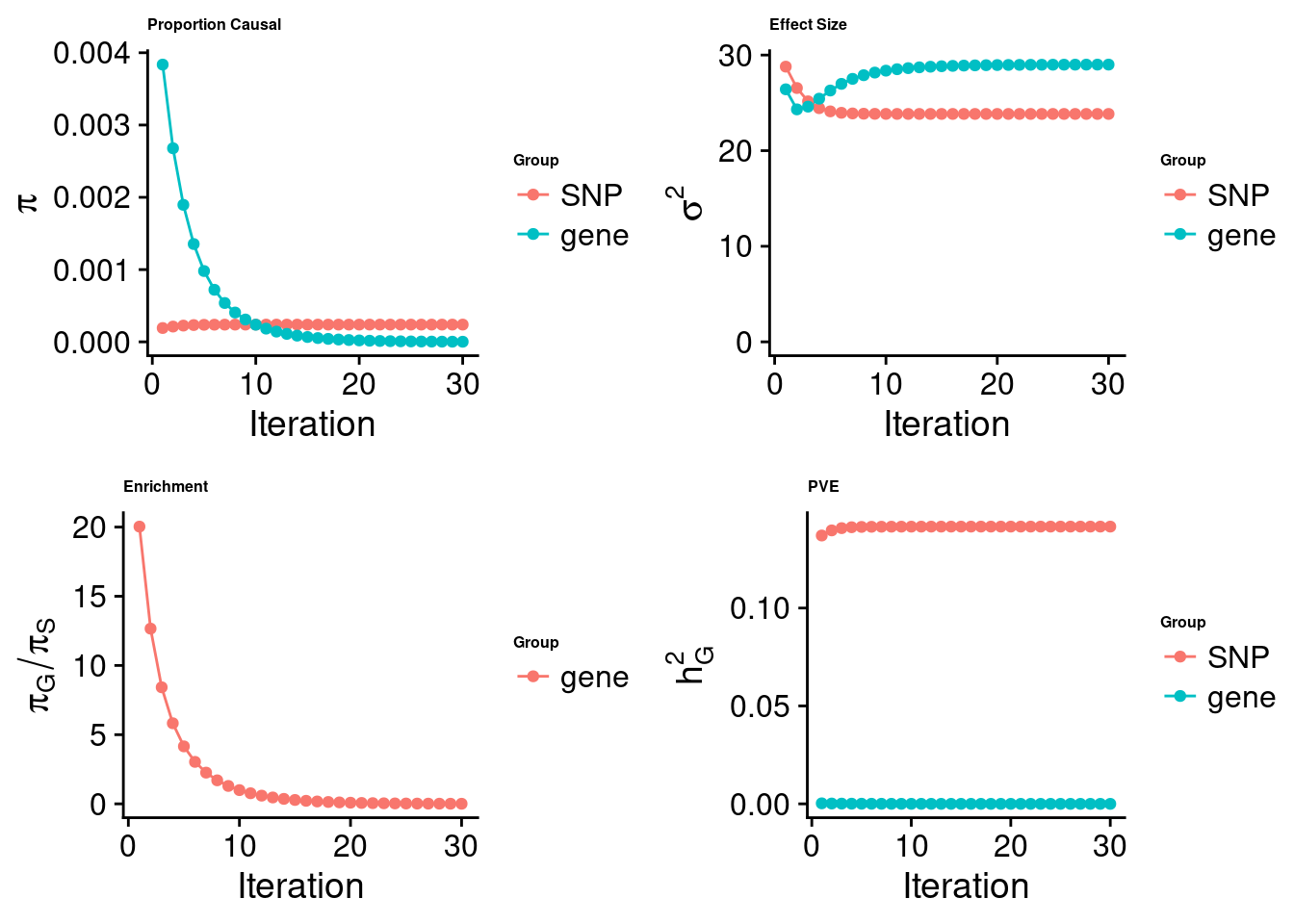
Joint analysis of expression, splicing and m6A
[1] "Check convergence for the top1 model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1928.0000 2123.000 888.0000
percent_of_overlaps 9.345e-01 0.9616 0.969 0.9673
SNP eQTL sQTL m6AQTL
estimated_group_prior 2.364e-04 0.019161 8.127e-03 3.872e-09
estimated_group_prior_var 2.553e+01 28.495736 1.868e+01 8.508e+00
estimated_group_pve 1.500e-01 0.003004 9.196e-04 8.348e-11
attributable_group_pve 9.745e-01 0.019509 5.973e-03 5.422e-10
[1] "Check convergence for the lasso model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1998.0000 2180.000 912.0000
percent_of_overlaps 9.345e-01 0.9965 0.995 0.9935
SNP eQTL sQTL m6AQTL
estimated_group_prior 2.143e-04 0.013424 2.429e-04 9.197e-11
estimated_group_prior_var 2.336e+01 62.619390 1.820e+01 2.083e+01
estimated_group_pve 1.245e-01 0.004792 2.749e-05 4.986e-12
attributable_group_pve 9.627e-01 0.037062 2.126e-04 3.856e-11$top1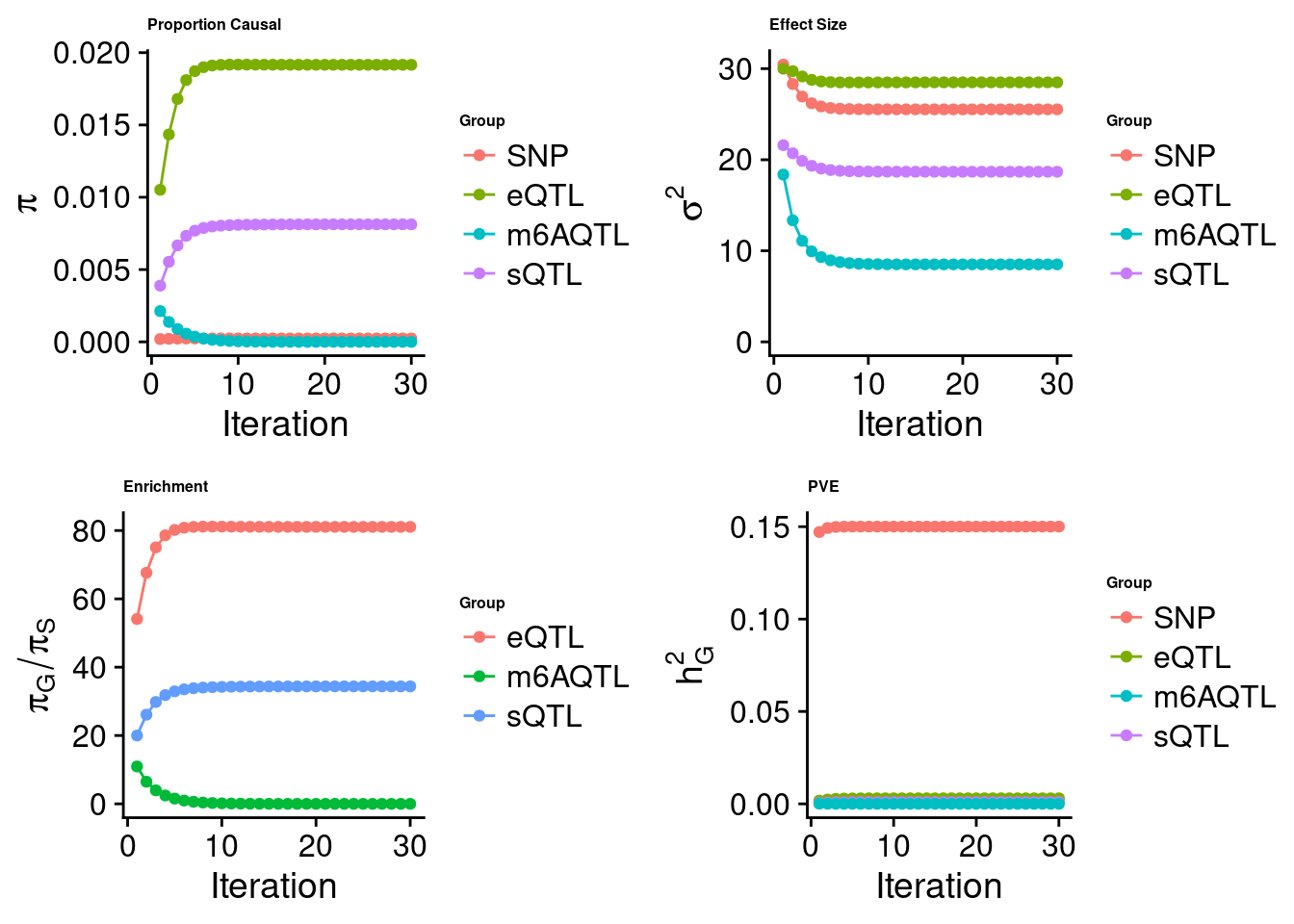
$lasso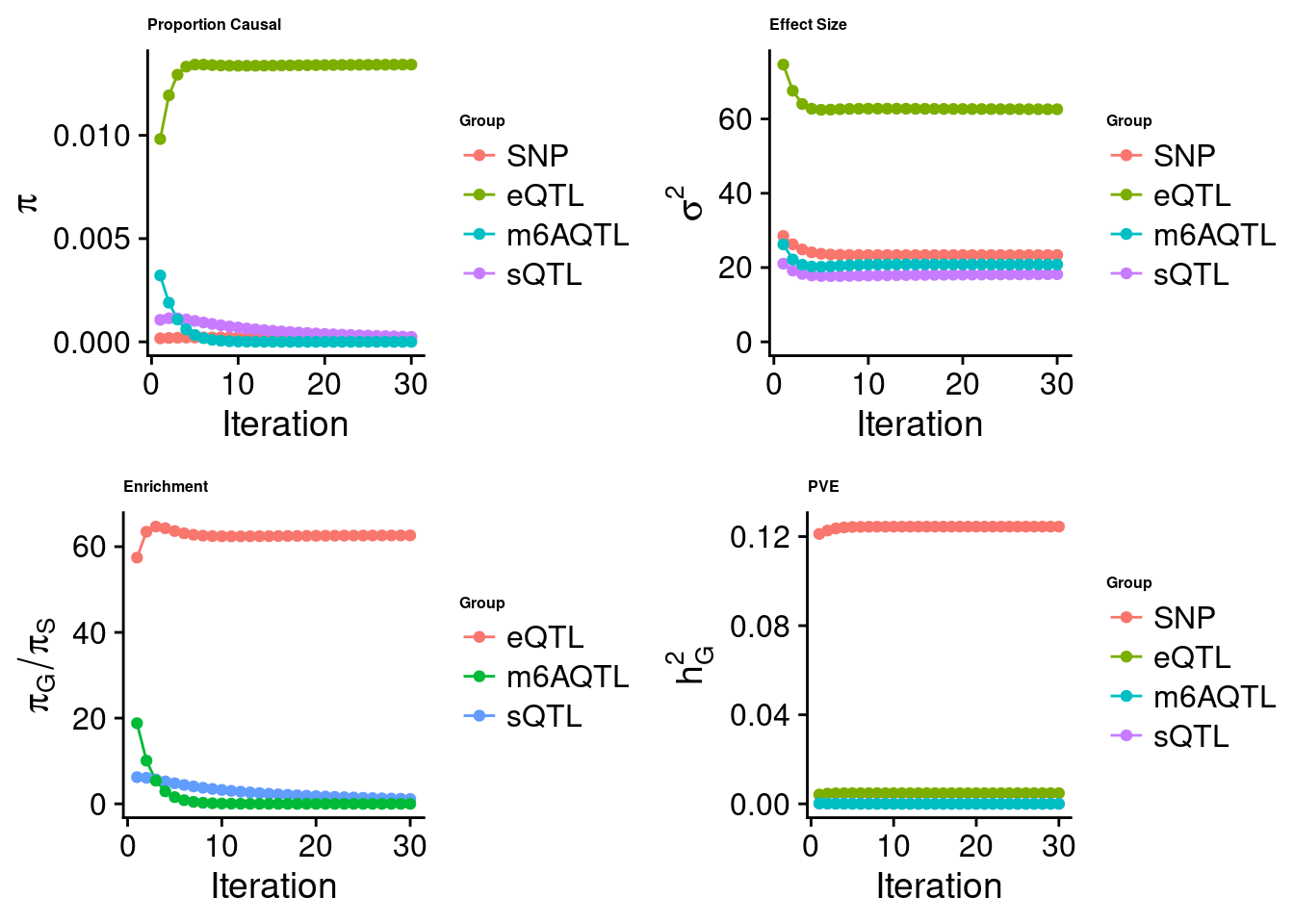
cTWAS results for individual analysis with m6A
Lasso model
[1] genename region_tag susie_pip z
<0 rows> (or 0-length row.names)Summing up PIPs for m6A peaks located in the same gene
Top m6A PIPs by genes
# A tibble: 0 × 2
# ℹ 2 variables: genename <chr>, total_susie_pip <dbl>cTWAS results for joint analysis using a lasso model
Top m6A modification pip
Top expression/splicing/m6A units
For m6A or splicing QTLs, they are assigned to the nearest genes (m6A needs to be confirmed with Kevin).
Top SNPs or genes with PIP > 0.6
$eQTL
genename susie_pip group region_tag
1914 ADAM15 0.9934 eQTL 1_79
1921 STK36 0.9925 eQTL 2_129
1995 ENSG00000205559 0.9801 eQTL 22_24
1924 SMIM19 0.9462 eQTL 8_37
1929 ENSG00000180139 0.8333 eQTL 10_57
1933 RBMS2 0.7782 eQTL 12_36
1527 SKAP1 0.7562 eQTL 17_28
1979 ELL 0.7499 eQTL 19_16
749 TSC22D4 0.6867 eQTL 7_61
1922 NSUN2 0.6741 eQTL 5_6
1202 GPN3 0.6468 eQTL 12_67
1368 CSNK1G1 0.6460 eQTL 15_29
912 PTPA 0.6099 eQTL 9_66
$m6AQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)
$sQTL
[1] genename susie_pip group region_tag
<0 rows> (or 0-length row.names)Top m6A modification pip
genename region_tag susie_pip z
1 TAP2 6_27 3.170e-08 -6.439
2 C2CD2L 11_71 1.818e-08 4.267
3 PSKH1 16_37 1.489e-08 -11.068
4 FERMT3 11_36 1.443e-08 -3.254
5 DHX38 16_38 1.261e-08 3.456
6 ZNF282 7_92 1.259e-08 3.636
7 ISG20 15_41 1.254e-08 -3.563
8 CD2BP2 16_24 9.926e-09 11.194
9 PARVB 22_19 7.998e-09 2.964
10 SMG9 19_30 6.096e-09 6.864Summing up PIPs for m6A peaks located in the same gene
Top 10 m6A PIPs by genes
# A tibble: 819 × 2
genename total_susie_pip
<chr> <dbl>
1 TAP2 0.0000000317
2 C2CD2L 0.0000000182
3 PSKH1 0.0000000149
4 FERMT3 0.0000000144
5 ISG20 0.0000000138
6 DHX38 0.0000000126
7 ZNF282 0.0000000126
8 CD2BP2 0.00000000993
9 PARVB 0.00000000800
10 SMG9 0.00000000610
# ℹ 809 more rowsTop splicing PIPs
peak_id genename pos region_tag susie_pip z
1 chr20:25204793-25205798 ENTPD6 25206654 20_19 0.31803 5.780
2 chr1:155679615-155686797 DAP3 155666961 1_79 0.03517 -1.429
3 chr11:66053043-66053172 YIF1A 65998757 11_37 0.03010 -2.807
4 chr15:75197572-75198619 FAM219B 75125645 15_35 0.02974 8.047
5 chr16:31503661-31504299 RUSF1 31470540 16_25 0.02597 -3.560
6 chr19:5717627-5719715 LONP1 5655792 19_5 0.02525 3.858
7 chr19:5714282-5719715 LONP1 5655792 19_5 0.02253 -3.822
8 chr8:38852924-38853732 TM2D2 38762056 8_35 0.02052 4.076
9 chr1:155658965-155664582 DAP3 155625602 1_79 0.01965 -1.188
10 chr1:93815558-93819465 DR1 93816400 1_57 0.01954 -6.505Summing up PIPs for spliced introns located in the same gene
Top 10 splicing PIPs by genes
# A tibble: 10 × 2
genename total_susie_pip
<chr> <dbl>
1 ENTPD6 0.318
2 DAP3 0.0553
3 LONP1 0.0478
4 DR1 0.0384
5 CIAO2A 0.0361
6 ENOSF1 0.0311
7 ATXN2L 0.0306
8 YIF1A 0.0304
9 FAM219B 0.0297
10 TRAF3IP3 0.0272Top genes by combined PIP
genename combined_pip expression_pip splicing_pip m6A_pip
81 ADAM15 0.993 0.9934 0.0000000 0.000e+00
2862 STK36 0.993 0.9925 0.0000000 0.000e+00
900 ENSG00000205559 0.980 0.9801 0.0000000 0.000e+00
2760 SMIM19 0.946 0.9462 0.0000000 0.000e+00
874 ENSG00000180139 0.833 0.8333 0.0000000 0.000e+00
2459 RBMS2 0.778 0.7782 0.0000000 0.000e+00
2687 SKAP1 0.760 0.7562 0.0037724 0.000e+00
826 ELL 0.750 0.7499 0.0000000 0.000e+00
3120 TSC22D4 0.687 0.6867 0.0000000 0.000e+00
2070 NSUN2 0.676 0.6741 0.0024136 0.000e+00
1420 GPN3 0.647 0.6468 0.0000000 0.000e+00
624 CSNK1G1 0.646 0.6460 0.0000000 4.822e-09
2386 PTPA 0.610 0.6099 0.0000000 0.000e+00
829 ELOA 0.579 0.5790 0.0000000 0.000e+00
2758 SMG9 0.572 0.5724 0.0000000 6.096e-09
460 CD22 0.569 0.5691 0.0001497 5.656e-11
306 BIK 0.552 0.5519 0.0000000 0.000e+00
1874 MPI 0.544 0.5259 0.0176415 0.000e+00
945 ENSG00000226526 0.519 0.5187 0.0000000 0.000e+00
665 DAP 0.500 0.4977 0.0018242 5.655e-10
region_tag
81 1_79
2862 2_129
900 22_24
2760 8_37
874 10_57
2459 12_36
2687 17_28
826 19_16
3120 7_61
2070 5_6
1420 12_67
624 15_29
2386 9_66
829 1_17
2758 19_30
460 19_25
306 22_18
1874 15_35
945 1_12
665 5_9Loading required package: gridWarning: replacing previous import 'utils::download.file' by
'restfulr::download.file' when loading 'rtracklayer'Locus plots for specific examples
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.52.0 Gviz_1.40.1 cowplot_1.1.1
[4] ggplot2_3.4.3 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[7] IRanges_2.30.1 S4Vectors_0.34.0 BiocGenerics_0.42.0
[10] ctwas_0.1.38 dplyr_1.1.2 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 deldir_1.0-6
[3] rjson_0.2.21 rprojroot_2.0.3
[5] biovizBase_1.44.0 htmlTable_2.4.0
[7] XVector_0.36.0 base64enc_0.1-3
[9] fs_1.6.3 dichromat_2.0-0.1
[11] rstudioapi_0.15.0 farver_2.1.1
[13] bit64_4.0.5 AnnotationDbi_1.58.0
[15] fansi_1.0.4 xml2_1.3.3
[17] codetools_0.2-18 logging_0.10-108
[19] cachem_1.0.8 knitr_1.39
[21] Formula_1.2-4 jsonlite_1.8.7
[23] Rsamtools_2.12.0 cluster_2.1.3
[25] dbplyr_2.3.3 png_0.1-7
[27] compiler_4.2.0 httr_1.4.6
[29] backports_1.4.1 lazyeval_0.2.2
[31] Matrix_1.6-1 fastmap_1.1.1
[33] cli_3.6.1 later_1.3.0
[35] htmltools_0.5.2 prettyunits_1.1.1
[37] tools_4.2.0 gtable_0.3.3
[39] glue_1.6.2 GenomeInfoDbData_1.2.8
[41] rappdirs_0.3.3 Rcpp_1.0.11
[43] Biobase_2.56.0 jquerylib_0.1.4
[45] vctrs_0.6.3 Biostrings_2.64.0
[47] rtracklayer_1.56.0 iterators_1.0.14
[49] xfun_0.30 stringr_1.5.0
[51] ps_1.7.0 lifecycle_1.0.3
[53] ensembldb_2.20.2 restfulr_0.0.14
[55] XML_3.99-0.14 getPass_0.2-2
[57] zlibbioc_1.42.0 scales_1.2.1
[59] BSgenome_1.64.0 VariantAnnotation_1.42.1
[61] ProtGenerics_1.28.0 hms_1.1.3
[63] promises_1.2.0.1 MatrixGenerics_1.8.0
[65] parallel_4.2.0 SummarizedExperiment_1.26.1
[67] AnnotationFilter_1.20.0 RColorBrewer_1.1-3
[69] yaml_2.3.5 curl_5.0.2
[71] memoise_2.0.1 gridExtra_2.3
[73] sass_0.4.1 rpart_4.1.16
[75] latticeExtra_0.6-30 stringi_1.7.12
[77] RSQLite_2.3.1 highr_0.9
[79] BiocIO_1.6.0 foreach_1.5.2
[81] checkmate_2.1.0 GenomicFeatures_1.48.4
[83] filelock_1.0.2 BiocParallel_1.30.3
[85] rlang_1.1.1 pkgconfig_2.0.3
[87] matrixStats_0.62.0 bitops_1.0-7
[89] evaluate_0.15 lattice_0.20-45
[91] htmlwidgets_1.5.4 GenomicAlignments_1.32.0
[93] labeling_0.4.2 bit_4.0.5
[95] processx_3.8.0 tidyselect_1.2.0
[97] magrittr_2.0.3 R6_2.5.1
[99] generics_0.1.3 Hmisc_5.1-0
[101] DelayedArray_0.22.0 DBI_1.1.3
[103] pgenlibr_0.3.6 pillar_1.9.0
[105] whisker_0.4 foreign_0.8-82
[107] withr_2.5.0 KEGGREST_1.36.2
[109] RCurl_1.98-1.7 nnet_7.3-17
[111] tibble_3.2.1 crayon_1.5.2
[113] interp_1.1-4 utf8_1.2.3
[115] BiocFileCache_2.4.0 rmarkdown_2.14
[117] jpeg_0.1-10 progress_1.2.2
[119] data.table_1.14.8 blob_1.2.4
[121] callr_3.7.3 git2r_0.30.1
[123] digest_0.6.33 httpuv_1.6.5
[125] munsell_0.5.0 bslib_0.3.1