cTWAS_analysis_for_white_blood_counts
Jing Gu
2023-08-16
Last updated: 2023-08-16
Checks: 6 1
Knit directory: m6A_in_disease_genetics/
This reproducible R Markdown analysis was created with workflowr (version 1.7.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20230331) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Using absolute paths to the files within your workflowr project makes it difficult for you and others to run your code on a different machine. Change the absolute path(s) below to the suggested relative path(s) to make your code more reproducible.
| absolute | relative |
|---|---|
| ~/projects/m6A_in_disease_genetics/code/ctwas/ctwas_config_b37.R | code/ctwas/ctwas_config_b37.R |
| ~/projects/m6A_in_disease_genetics/code/ctwas/qiansheng/locus_plot.R | code/ctwas/qiansheng/locus_plot.R |
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 08eaf44. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .ipynb_checkpoints/
Ignored: analysis/m6A_switch_to_disease_h2g.nb.html
Ignored: data/plots/
Untracked files:
Untracked: HMGCR_locus_gene_tracks.pdf
Untracked: Rplots.pdf
Untracked: analysis/.ipynb_checkpoints/
Untracked: analysis/IBD_E_S_m6A.Rmd
Untracked: analysis/IBD_E_S_m6A_output.Rmd
Untracked: analysis/LDL_E_S_m6A.Rmd
Untracked: analysis/LDL_m6A_output.Rmd
Untracked: analysis/RA_m6A_output.Rmd
Untracked: analysis/WhiteBlood_WholeBlood_E_M.Rmd
Untracked: analysis/identify_m6A_mechanisms_with_finemapping.Rmd
Untracked: analysis/lymph_m6A_output.Rmd
Untracked: analysis/pre_weights_m6AQTL.txt
Untracked: analysis/rbc_E_S_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output.Rmd
Untracked: analysis/rbc_m6A_output_hg19.Rmd
Untracked: analysis/wbc_E_S_m6A_output.Rmd
Untracked: code/.ipynb_checkpoints/
Untracked: code/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: code/check_double_strand.ipynb
Untracked: code/check_double_strand_v2.ipynb
Untracked: code/ctwas/
Untracked: code/figure/
Untracked: code/learn_gviz.Rmd
Untracked: code/learn_gviz.html
Untracked: code/learn_gviz.nb.html
Untracked: code/m6AQTL_finemapping.Rmd
Untracked: code/summary_TWAS_coloc_m6A_2023.Rmd
Untracked: code/test_gviz.ipynb
Untracked: code/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: data/.ipynb_checkpoints/
Untracked: data/ADCY7_gwas_input.tsv
Untracked: data/ADCY7_qtl_input.tsv
Untracked: data/Allergy_full_coloc.txt
Untracked: data/Asthma_full_coloc.txt
Untracked: data/CAD_full_coloc.txt
Untracked: data/Eosinophil_count_full_coloc.txt
Untracked: data/GSE125377_jointPeakReadCount.txt
Untracked: data/HMGCR_ctwas_dat.Rd
Untracked: data/IBD_full_coloc.txt
Untracked: data/JointPeaks.bed
Untracked: data/Li2022_dsRNAs.xlsx
Untracked: data/Lupus_full_coloc.txt
Untracked: data/RA_full_coloc.txt
Untracked: data/TABLE1_hg19.txt
Untracked: data/TABLE1_hg19.txt.zip
Untracked: data/__MACOSX/
Untracked: data/coloc_blood_traits.csv
Untracked: data/crohns_disease_full_coloc.txt
Untracked: data/edit_sites_and_GE_neg_correlated.txt
Untracked: data/edit_sites_and_GE_pos_correlated.txt
Untracked: data/features
Untracked: data/human_EERs.csv
Untracked: data/human_EERs.txt
Untracked: data/lymph_full_coloc.txt
Untracked: data/m6A_TWAS_results.csv
Untracked: data/m6a_TWAS_genes.txt
Untracked: data/m6a_joint_calling_peaks.csv
Untracked: data/nasser_2021_ABC_IBD_genes.txt
Untracked: data/nat_sense_pairs.csv
Untracked: data/plt_full_coloc.txt
Untracked: data/rbc_full_coloc.txt
Untracked: data/rdw_full_coloc.txt
Untracked: data/reported_AS_targets_S1.txt
Untracked: data/reported_AS_wanowska.txt
Untracked: data/sig_coloc_results/
Untracked: data/test_locuscomparer.pdf
Untracked: data/ulcerative_colitis_full_coloc.txt
Untracked: data/wbc_full_coloc.txt
Untracked: data/zhao_silver_genes.csv
Untracked: output/.ipynb_checkpoints/
Untracked: output/HMGCR_gene_track_plot.pdf
Untracked: output/HMGCR_locus_plot.pdf
Untracked: output/all_m6a_sites_with_cisNATs.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.3.csv
Untracked: output/all_m6a_sites_with_paired_cisNATs_summary_PP40.5.csv
Untracked: output/all_m6a_sites_with_paired_cis_NATs.csv
Untracked: output/fine_mapped_m6AQTLs_TWAS_genes_highPP4.rds
Untracked: output/gene_summary.csv
Untracked: output/immune_related_m6A_targets.csv
Untracked: output/m6aQTL_dsRNAs_PPP2R3C_PRORP.pdf
Untracked: output/m6a_peaks_nearby_dsRNAs.csv
Untracked: output/m6a_sites_near_all_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_coloc.csv
Untracked: output/m6a_sites_near_dsRNAs_twas.csv
Untracked: output/m6a_sites_near_dsRNAs_twas_summary.csv
Untracked: output/m6a_sites_overlapping_NAT_twas.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_coloc.csv
Untracked: output/m6a_sites_overlapping_dsRNAs_twas.csv
Untracked: output/m6a_sites_overlapping_dsRegions.csv
Untracked: output/m6a_sites_overlapping_dsRegions_coloc.csv
Untracked: output/negatively_correlated_genes.txt
Untracked: output/postively_correlated_genes.txt
Untracked: output/rs1806261_RABEP1-NUP88_focused_locusview.pdf
Untracked: output/rs1806261_RABEP1-NUP88_locusview.pdf
Untracked: output/rs3177647_MAPKAPK5-AS1-MAPKAPK5_locusview.pdf
Untracked: output/rs3204541_DDX55-EIF2B1_locusview.pdf
Untracked: output/rs7184802_ADCY7-BRD7_locusview.pdf
Untracked: output/rs7184802_ADCY7_locuscompare.pdf
Untracked: output/twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Untracked: output/twas_genes_PP4_0.5_blood_traits_trackplots.pdf
Untracked: output/twas_m6a_sites_with_all_cisNATs.RDS
Untracked: output/twas_m6a_sites_with_cisNATs_range.RDS
Untracked: output/twas_m6a_sites_with_the_nearest_cisNAT.RDS
Untracked: twas_genes_PP4_0.3_immune_traits_trackplots.pdf
Unstaged changes:
Modified: analysis/m6A_switch_to_disease_h2g.Rmd
Modified: analysis/wbc_m6A_output.Rmd
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/wbc_m6A_output_hg19.Rmd)
and HTML (docs/wbc_m6A_output_hg19.html) files. If you’ve
configured a remote Git repository (see ?wflow_git_remote),
click on the hyperlinks in the table below to view the files as they
were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 08eaf44 | Jing Gu | 2023-08-16 | run ctwas for multiple traits |
| html | 3a4bab9 | Jing Gu | 2023-08-11 | Build site. |
| Rmd | d1d6b2a | Jing Gu | 2023-08-11 | wflow_publish(c("analysis/wbc_m6A_output_hg19.Rmd", "analysis/index.Rmd", |
| Rmd | c94ee10 | Jing Gu | 2023-08-11 | wflow_publish(c("analysis/wbc_m6A_output_hg19.Rmd", "analysis/index.Rmd", |
Load ctwas results
# top 1 method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "recover",
recover_strand_ambig_wgt = T
# lasso/elastic-net method
res <- impute_expr_z(z_snp, weight = weight, ld_R_dir = ld_R_dir,
method = NULL, outputdir = outputdir, outname = outname.e,
harmonize_z = T, harmonize_wgt = T, scale_by_ld_variance=F,
strand_ambig_action_z = "none",
recover_strand_ambig_wgt = FGWAS: UK Biobank GWAS summary statistics - European individuals
Weights: FUSION weights using top1, lasso, or elastic-net models were converted into PredictDB format and were not needed to do scaling when running ctwas.
Check convergence of parameters
cTWAS analysis on m6A alone
[1] "Check convergence for the top1 model:"
[1] "Table of group size:"
SNP gene
8713250 888
SNP gene
estimated_group_prior 2.481e-04 1.227e-02
estimated_group_prior_var 1.920e+01 2.631e+01
estimated_group_pve 1.184e-01 8.178e-04
attributable_group_pve 9.931e-01 6.858e-03
[1] "Check convergence for the lasso model:"
[1] "Table of group size:"
SNP gene
8713250 912
SNP gene
estimated_group_prior 2.414e-04 1.016e-02
estimated_group_prior_var 1.898e+01 3.699e+01
estimated_group_pve 1.139e-01 9.778e-04
attributable_group_pve 9.915e-01 8.513e-03$top1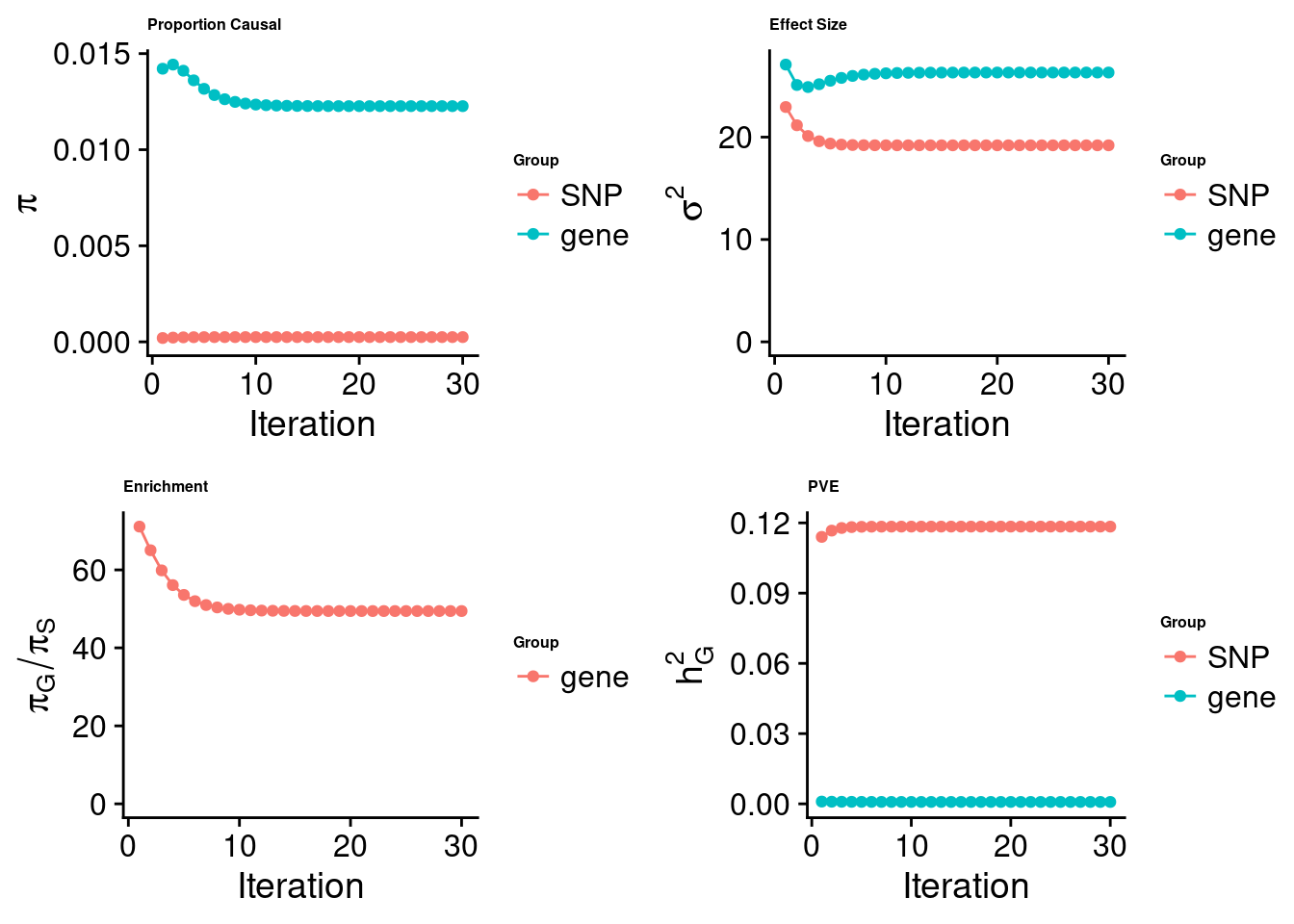
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
$lasso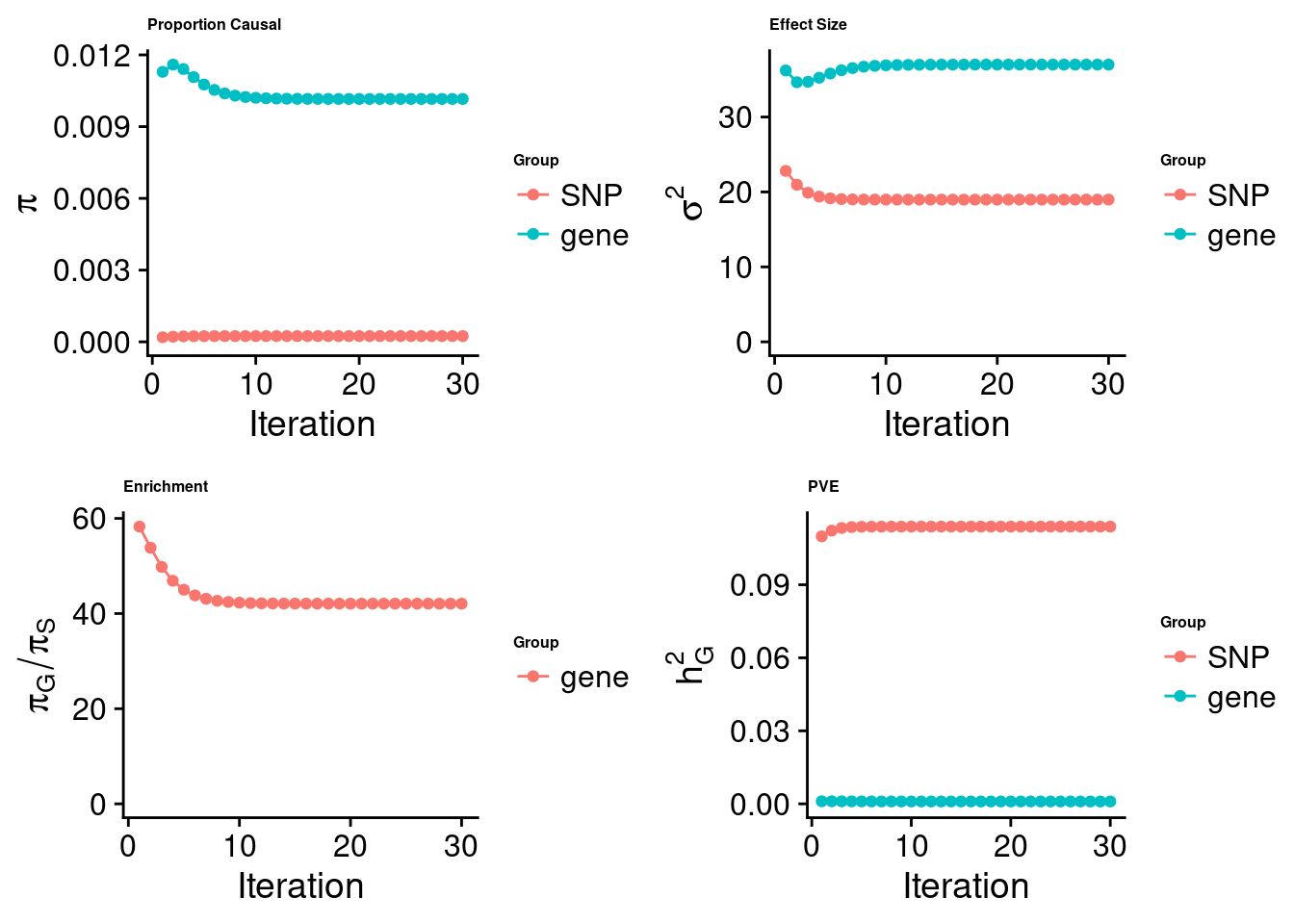
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
Joint analysis of expression, splicing and m6A
[1] "Check convergence for the top1 model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1928.0000 2123.000 888.0000
percent_of_overlaps 9.345e-01 0.9616 0.969 0.9673
SNP eQTL sQTL m6AQTL
estimated_group_prior 2.406e-04 8.895e-03 0.012934 1.236e-02
estimated_group_prior_var 1.858e+01 1.683e+01 36.589120 2.554e+01
estimated_group_pve 1.112e-01 8.236e-04 0.002867 7.999e-04
attributable_group_pve 9.612e-01 7.120e-03 0.024783 6.916e-03
[1] "Check convergence for the lasso model when jointly analyzing expression, splicing and m6A:"
[1] "Table of group size before/after matching with UKBB SNPs:"
SNP eQTL sQTL m6AQTL
prior_group_size 9.324e+06 2005.0000 2191.000 918.0000
group_size 8.713e+06 1998.0000 2180.000 912.0000
percent_of_overlaps 9.345e-01 0.9965 0.995 0.9935
SNP eQTL sQTL m6AQTL
estimated_group_prior 0.000217 9.483e-04 0.005262 0.012026
estimated_group_prior_var 19.281964 2.155e+01 38.383079 33.143952
estimated_group_pve 0.104003 1.165e-04 0.001256 0.001037
attributable_group_pve 0.977353 1.095e-03 0.011805 0.009747$top1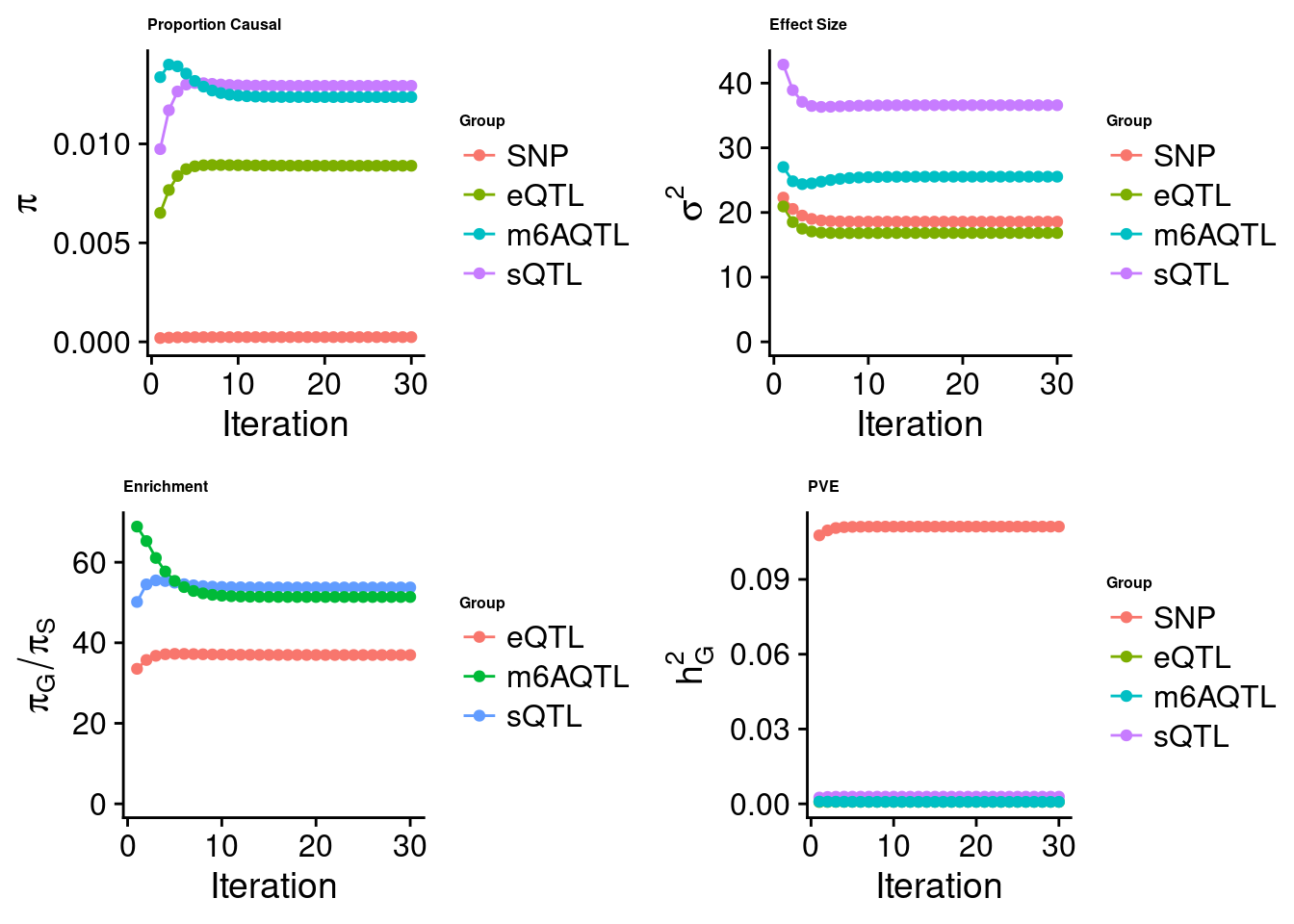
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
$lasso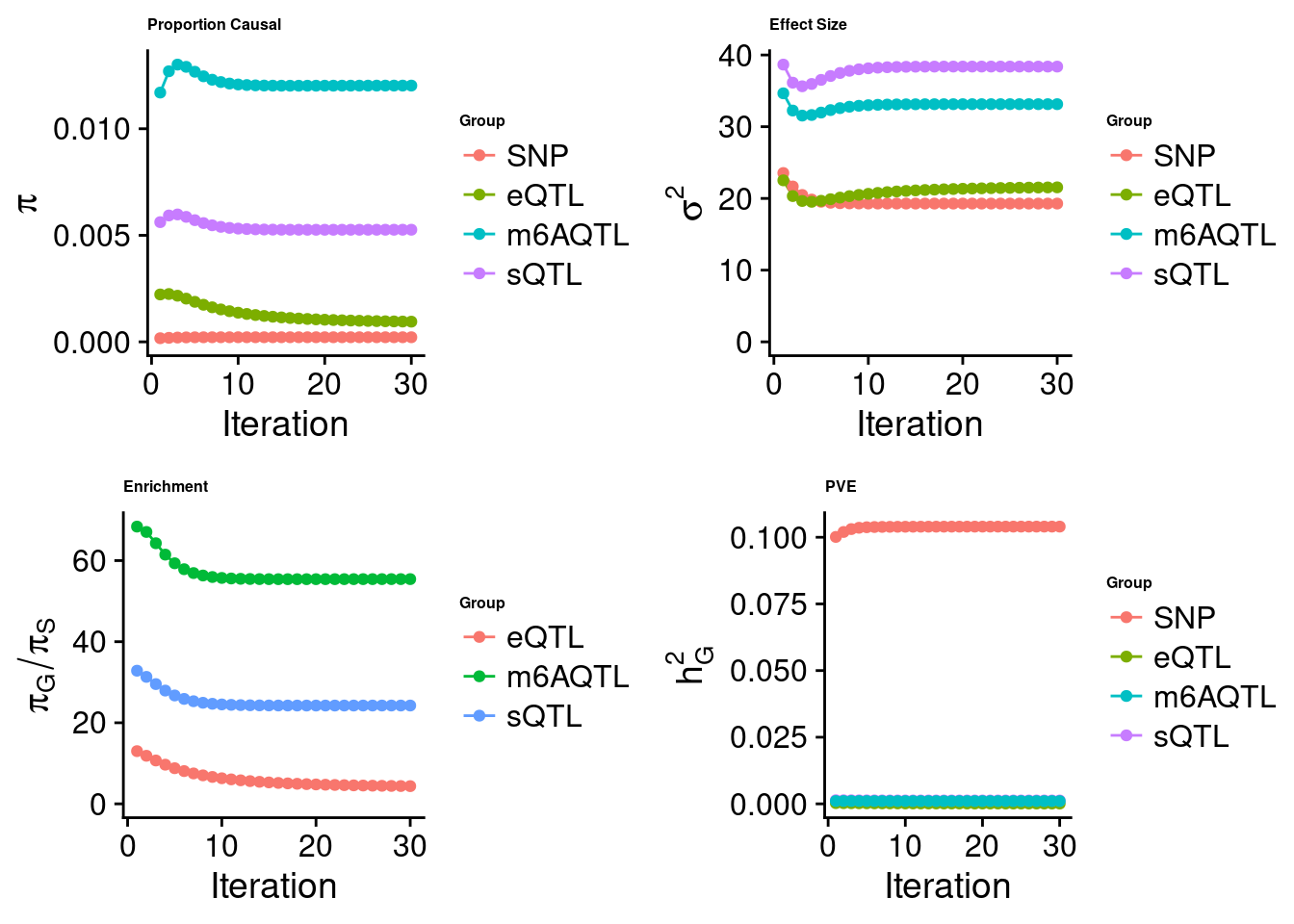
cTWAS results for individual analysis with m6A
Lasso model
genename region_tag susie_pip z
1 SLC9A3R1 17_42 0.9473 -7.630
2 ZKSCAN5 7_61 0.7976 7.112
3 ADCY7 16_27 0.7817 4.382
4 TRIT1 1_25 0.7516 5.554
5 THEMIS2 1_19 0.7034 6.243
6 BTN3A3 6_20 0.6855 -13.445
7 WAC-AS1 10_20 0.6102 11.178Summing up PIPs for m6A peaks located in the same gene
Top m6A PIPs by genes
# A tibble: 7 × 2
genename total_susie_pip
<chr> <dbl>
1 SLC9A3R1 0.947
2 ZKSCAN5 0.798
3 ADCY7 0.782
4 TRIT1 0.752
5 THEMIS2 0.703
6 BTN3A3 0.686
7 WAC-AS1 0.615cTWAS results for joint analysis using a lasso model
Top m6A modification pip
Top expression/splicing/m6A units
For m6A or splicing QTLs, they are assigned to the nearest genes (m6A needs to be confirmed with Kevin).
Top SNPs or genes with PIP > 0.6
$eQTL
genename susie_pip group region_tag
1981 CSNK1G1 0.9988 eQTL 15_29
43 AL391650.1 0.7867 eQTL 1_18
143 NDUFS2 0.7360 eQTL 1_81
1915 TTLL12 0.6001 eQTL 22_18
$m6AQTL
genename susie_pip group region_tag
5089 SLC9A3R1 0.9586 m6AQTL 17_42
5072 ZKSCAN5 0.8403 m6AQTL 7_61
4202 TRIT1 0.7973 m6AQTL 1_25
4196 THEMIS2 0.7560 m6AQTL 1_19
4425 BTN3A3 0.7494 m6AQTL 6_20
5084 ADCY7 0.7069 m6AQTL 16_27
4529 DENND3 0.6867 m6AQTL 8_92
4586 WAC-AS1 0.6349 m6AQTL 10_20
$sQTL
genename susie_pip group region_tag
4119 RNF181 1.0000 sQTL 2_54
4126 HLA-F 1.0000 sQTL 6_23
4136 MYO1G 0.9983 sQTL 7_33
4164 FNBP4 0.9889 sQTL 11_29
2471 GSK3B 0.6478 sQTL 3_74
3084 PDLIM1 0.6441 sQTL 10_61Top m6A modification pip
ZKSCAN5: RNA Polymerase II Cis-Regulatory Region Sequence-Specific DNA Binding (GO:0000978). THEMIS2 is involved in the biological process T Cell Receptor Signaling Pathway (GO:0050852). BANF: DNA binding factor|Regulation Of Innate Immune Response (GO:0045088). TRIT1 has the molecular function of Catalytic Activity, Acting On A tRNA (GO:0140101). TRIT1 is involved in the biological process RNA Modification (GO:0009451). S1PR2 is involved in the biological process Regulation Of Cell Population Proliferation (GO:0042127). WAC has the molecular function of RNA Polymerase II Complex Binding (GO:0000993). CD320 is involved in the biological process Regulation Of B Cell Proliferation (GO:0030888).
genename region_tag susie_pip z
1 SLC9A3R1 17_42 0.9586 -7.630
2 ZKSCAN5 7_61 0.8403 7.112
3 TRIT1 1_25 0.7973 5.554
4 THEMIS2 1_19 0.7560 6.243
5 BTN3A3 6_20 0.7494 -13.445
6 ADCY7 16_27 0.7069 4.382
7 DENND3 8_92 0.6867 5.979
8 WAC-AS1 10_20 0.6349 11.178
9 SMG9 19_30 0.5570 4.092
10 SQSTM1 5_108 0.4403 -4.393Summing up PIPs for m6A peaks located in the same gene
Top 10 m6A PIPs by genes
# A tibble: 819 × 2
genename total_susie_pip
<chr> <dbl>
1 SLC9A3R1 0.959
2 ZKSCAN5 0.840
3 TRIT1 0.797
4 THEMIS2 0.756
5 BTN3A3 0.749
6 ADCY7 0.707
7 DENND3 0.687
8 WAC-AS1 0.642
9 SMG9 0.557
10 SQSTM1 0.440
# ℹ 809 more rowsTop splicing PIPs
Some loci contain variants in the same credible set but having opposite z scores. For instance, the predicted splicing levels of two introns of CNN2 based on the same variant (position=1038445) have opposite associations with traits. Is this variant more likely to affect traits by altering the splicing levels of both transcripts, rather than one of them since they have equal PIP?
peak_id genename pos region_tag susie_pip z
1 chr2:85823772-85824227 RNF181 85818886 2_54 1.0000 5.175
2 chr6:29693340-29694660 HLA-F 29646165 6_23 1.0000 -17.265
3 chr7:45009474-45009639 MYO1G 44925489 7_33 0.9983 -11.848
4 chr11:47761655-47765505 FNBP4 47684908 11_29 0.9889 10.996
5 chr3:119582452-119624602 GSK3B 119503971 3_74 0.6478 5.631
6 chr10:97007123-97023621 PDLIM1 97001124 10_61 0.6441 -7.375
7 chr9:86593367-86595418 HNRNPK 86592026 9_41 0.5989 9.019
8 chr22:24268707-24316496 AC253536.7 24182500 22_7 0.5457 6.311
9 chr1:43852637-43853174 MED8 43843649 1_27 0.4548 4.806
10 chr5:122111457-122130961 SNX2 122050927 5_74 0.4518 -6.744Summing up PIPs for spliced introns located in the same gene
Top 10 splicing PIPs by genes
# A tibble: 10 × 2
genename total_susie_pip
<chr> <dbl>
1 HLA-F 1.00
2 RNF181 1
3 MYO1G 0.998
4 FNBP4 0.989
5 HNRNPK 0.910
6 MED8 0.886
7 CNN2 0.852
8 CD46 0.725
9 GSK3B 0.648
10 PDLIM1 0.644Top genes by combined PIP
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
624 CSNK1G1 1.018 9.988e-01 0.000000 1.903e-02 15_29
1505 HLA-F 1.000 1.021e-08 1.000037 3.634e-07 6_23
2517 RNF181 1.000 0.000e+00 1.000000 0.000e+00 2_54
1957 MYO1G 0.998 0.000e+00 0.998301 0.000e+00 7_33
1340 FNBP4 0.989 0.000e+00 0.988933 0.000e+00 11_29
2741 SLC9A3R1 0.959 0.000e+00 0.000000 9.586e-01 17_42
1518 HNRNPK 0.937 0.000e+00 0.910055 2.663e-02 9_41
1807 MED8 0.886 0.000e+00 0.885964 0.000e+00 1_27
567 CNN2 0.852 0.000e+00 0.852357 0.000e+00 19_2
3317 ZKSCAN5 0.840 0.000e+00 0.000000 8.403e-01 7_61
3103 TRIT1 0.799 4.314e-05 0.001363 7.973e-01 1_25
129 AL391650.1 0.787 7.867e-01 0.000000 0.000e+00 1_18
2962 THEMIS2 0.756 0.000e+00 0.000000 7.560e-01 1_19
339 BTN3A3 0.751 1.570e-03 0.000000 7.494e-01 6_20
2012 NDUFS2 0.736 7.360e-01 0.000000 0.000e+00 1_81
471 CD46 0.725 0.000e+00 0.724809 0.000e+00 1_107
700 DENND3 0.724 0.000e+00 0.037520 6.867e-01 8_92
86 ADCY7 0.707 0.000e+00 0.000000 7.069e-01 16_27
1439 GSK3B 0.648 0.000e+00 0.647811 0.000e+00 3_74
2184 PDLIM1 0.644 0.000e+00 0.644131 0.000e+00 10_61Loading required package: gridWarning: replacing previous import 'utils::download.file' by
'restfulr::download.file' when loading 'rtracklayer'Locus plots for specific examples
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
2012 NDUFS2 0.736 0.736 0 0 1_81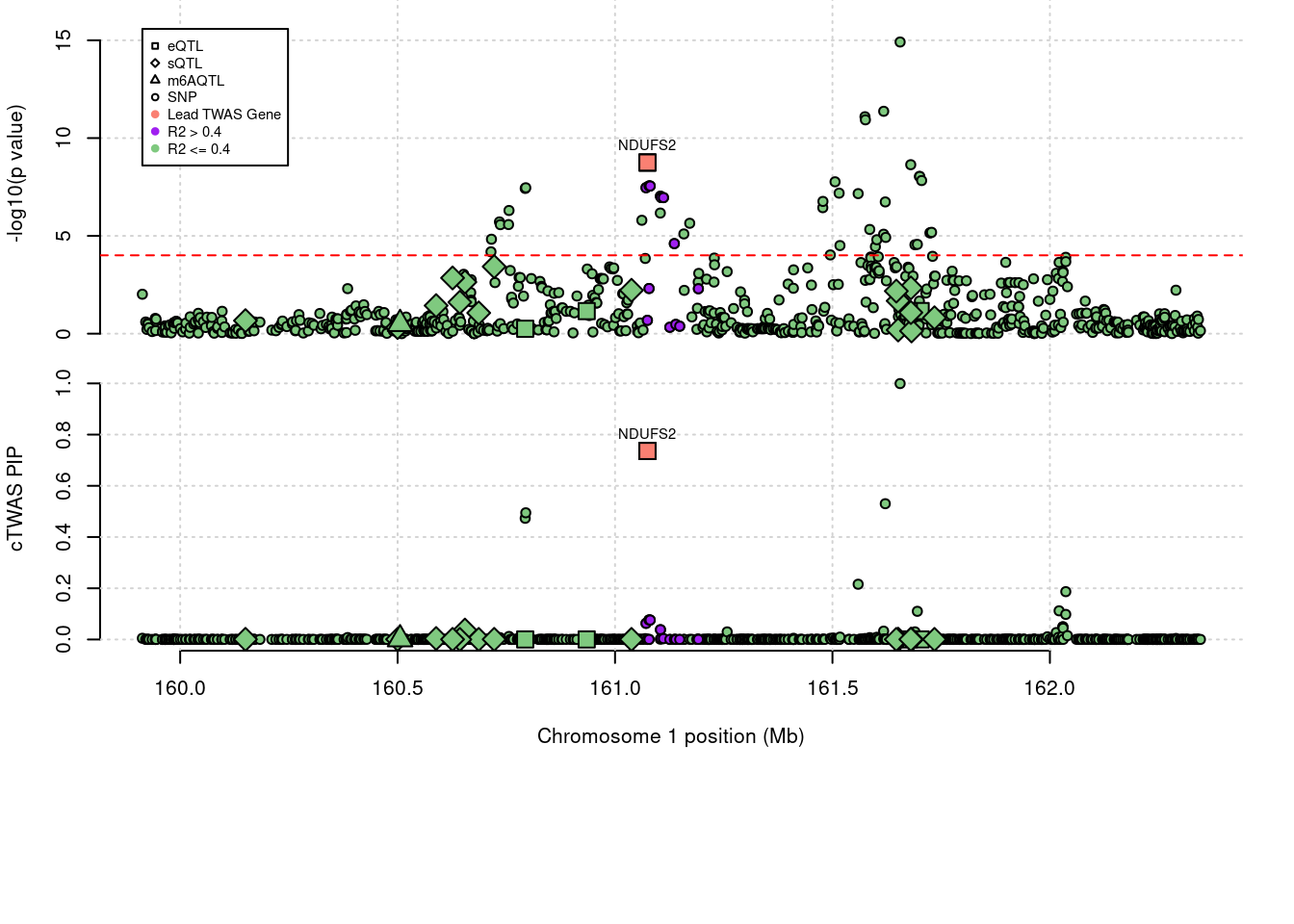
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
624 CSNK1G1 1.018 0.9988 0 0.01903 15_29Warning in asMethod(object): sparse->dense coercion: allocating vector of size
1.1 GiB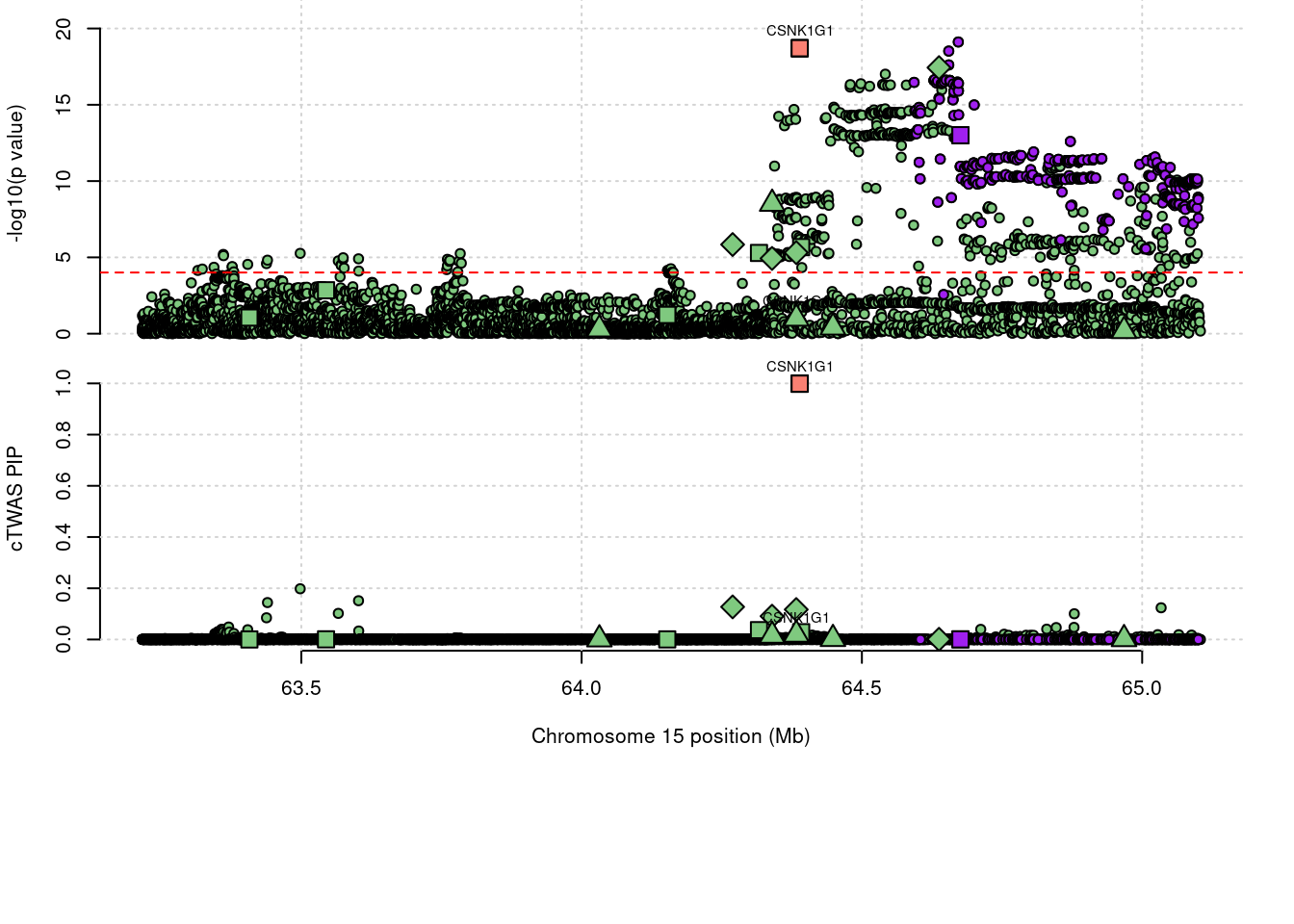
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
2962 THEMIS2 0.756 0 0 0.756 1_19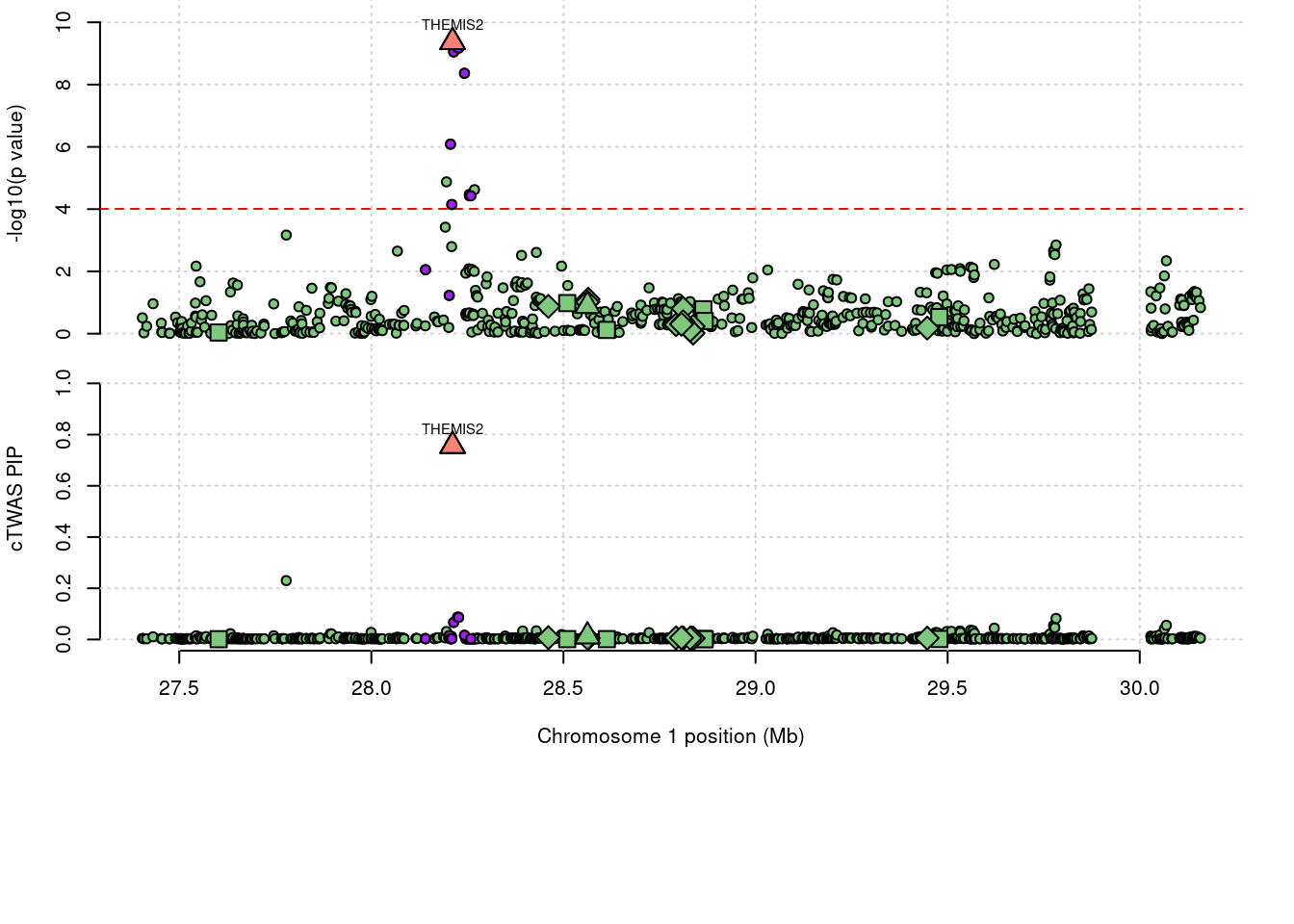
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
genename combined_pip expression_pip splicing_pip m6A_pip region_tag
3317 ZKSCAN5 0.84 0 0 0.8403 7_61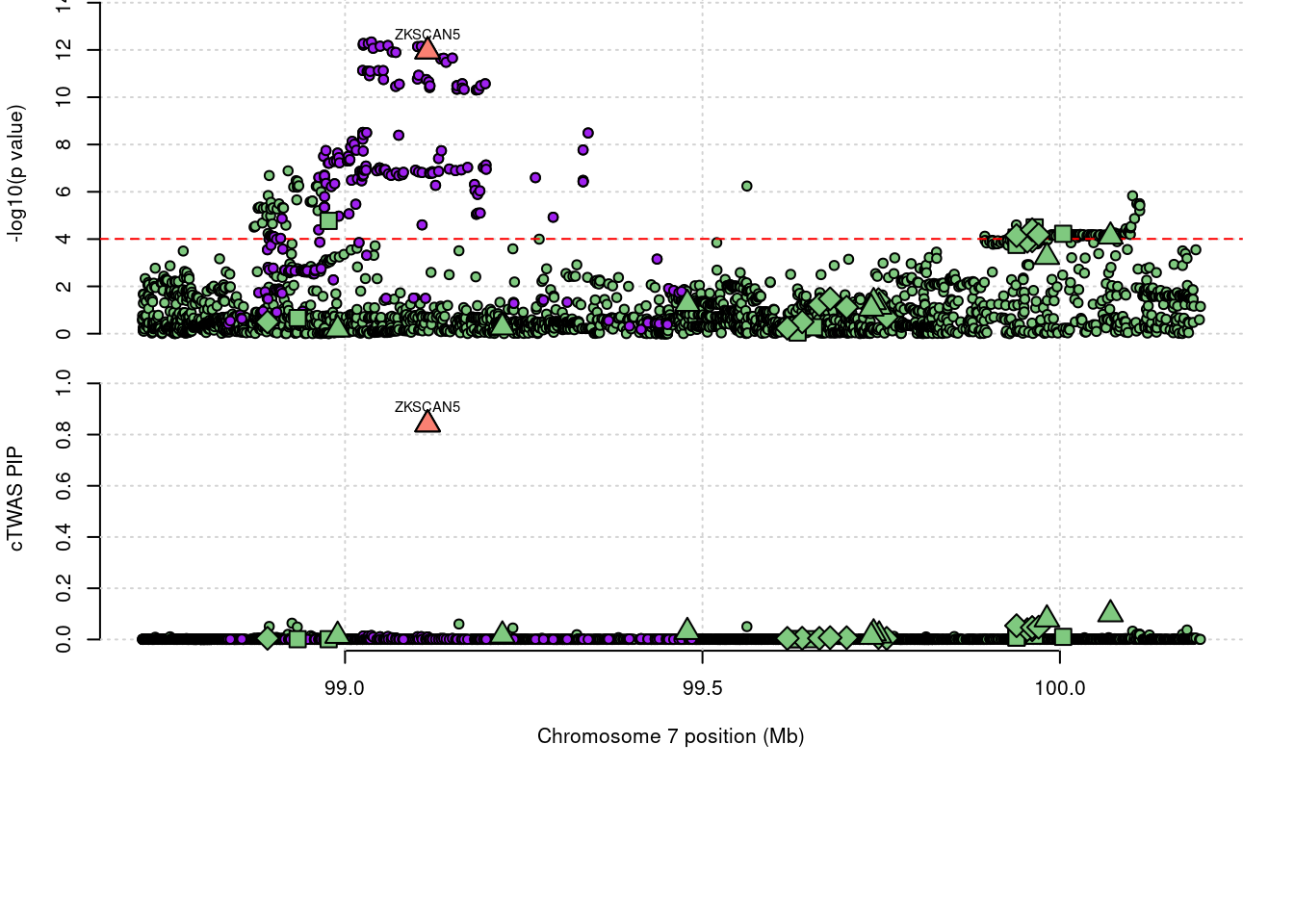
| Version | Author | Date |
|---|---|---|
| 3a4bab9 | Jing Gu | 2023-08-11 |
R version 4.2.0 (2022-04-22)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: CentOS Linux 7 (Core)
Matrix products: default
BLAS/LAPACK: /software/openblas-0.3.13-el7-x86_64/lib/libopenblas_haswellp-r0.3.13.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=C
[4] LC_COLLATE=C LC_MONETARY=C LC_MESSAGES=C
[7] LC_PAPER=C LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] biomaRt_2.52.0 Gviz_1.40.1 cowplot_1.1.1
[4] ggplot2_3.4.3 GenomicRanges_1.48.0 GenomeInfoDb_1.32.2
[7] IRanges_2.30.1 S4Vectors_0.34.0 BiocGenerics_0.42.0
[10] ctwas_0.1.38 dplyr_1.1.2 workflowr_1.7.0
loaded via a namespace (and not attached):
[1] colorspace_2.1-0 deldir_1.0-6
[3] rjson_0.2.21 rprojroot_2.0.3
[5] biovizBase_1.44.0 htmlTable_2.4.0
[7] XVector_0.36.0 base64enc_0.1-3
[9] fs_1.6.3 dichromat_2.0-0.1
[11] rstudioapi_0.15.0 farver_2.1.1
[13] bit64_4.0.5 AnnotationDbi_1.58.0
[15] fansi_1.0.4 xml2_1.3.3
[17] codetools_0.2-18 logging_0.10-108
[19] cachem_1.0.8 knitr_1.39
[21] Formula_1.2-4 jsonlite_1.8.7
[23] Rsamtools_2.12.0 cluster_2.1.3
[25] dbplyr_2.3.3 png_0.1-7
[27] compiler_4.2.0 httr_1.4.6
[29] backports_1.4.1 lazyeval_0.2.2
[31] Matrix_1.6-1 fastmap_1.1.1
[33] cli_3.6.1 later_1.3.0
[35] htmltools_0.5.2 prettyunits_1.1.1
[37] tools_4.2.0 gtable_0.3.3
[39] glue_1.6.2 GenomeInfoDbData_1.2.8
[41] rappdirs_0.3.3 Rcpp_1.0.11
[43] Biobase_2.56.0 jquerylib_0.1.4
[45] vctrs_0.6.3 Biostrings_2.64.0
[47] rtracklayer_1.56.0 iterators_1.0.14
[49] xfun_0.30 stringr_1.5.0
[51] ps_1.7.0 lifecycle_1.0.3
[53] ensembldb_2.20.2 restfulr_0.0.14
[55] XML_3.99-0.14 getPass_0.2-2
[57] zlibbioc_1.42.0 scales_1.2.1
[59] BSgenome_1.64.0 VariantAnnotation_1.42.1
[61] ProtGenerics_1.28.0 hms_1.1.3
[63] promises_1.2.0.1 MatrixGenerics_1.8.0
[65] parallel_4.2.0 SummarizedExperiment_1.26.1
[67] AnnotationFilter_1.20.0 RColorBrewer_1.1-3
[69] yaml_2.3.5 curl_5.0.2
[71] memoise_2.0.1 gridExtra_2.3
[73] sass_0.4.1 rpart_4.1.16
[75] latticeExtra_0.6-30 stringi_1.7.12
[77] RSQLite_2.3.1 highr_0.9
[79] BiocIO_1.6.0 foreach_1.5.2
[81] checkmate_2.1.0 GenomicFeatures_1.48.4
[83] filelock_1.0.2 BiocParallel_1.30.3
[85] rlang_1.1.1 pkgconfig_2.0.3
[87] matrixStats_0.62.0 bitops_1.0-7
[89] evaluate_0.15 lattice_0.20-45
[91] htmlwidgets_1.5.4 GenomicAlignments_1.32.0
[93] labeling_0.4.2 bit_4.0.5
[95] processx_3.8.0 tidyselect_1.2.0
[97] magrittr_2.0.3 R6_2.5.1
[99] generics_0.1.3 Hmisc_5.1-0
[101] DelayedArray_0.22.0 DBI_1.1.3
[103] pgenlibr_0.3.6 pillar_1.9.0
[105] whisker_0.4 foreign_0.8-82
[107] withr_2.5.0 KEGGREST_1.36.2
[109] RCurl_1.98-1.7 nnet_7.3-17
[111] tibble_3.2.1 crayon_1.5.2
[113] interp_1.1-4 utf8_1.2.3
[115] BiocFileCache_2.4.0 rmarkdown_2.14
[117] jpeg_0.1-10 progress_1.2.2
[119] data.table_1.14.8 blob_1.2.4
[121] callr_3.7.3 git2r_0.30.1
[123] digest_0.6.33 httpuv_1.6.5
[125] munsell_0.5.0 bslib_0.3.1